
Abstract
It is clear that the immune system and inflammation have a role in Parkinson’s disease (PD), including sporadic PD and some genetic forms such as LRRK2-associated PD. One of the most important genes associated with PD is GBA1, as variants in this gene are found in 5–20% of PD patients in different populations worldwide. Biallelic variants in GBA1 may cause Gaucher disease, a lysosomal storage disorder with involvement of the immune system, and other lines of evidence link GBA1 to the immune system and inflammation. In this review, we discuss these different pieces of evidence and whether the interplay between GBA1 and the immune system may have a role in PD.
INTRODUCTION
The immune system and inflammation seem to have an important role in Parkinson’s disease (PD), with multiple lines of evidence from pathological, genetic, serological, and experimental studies [1]. From the genetic perspective, the association with specific human leukocyte antigen (HLA) types and amino acid changes [2, 3] and with immune-related genes including LRRK2 and BST1 [4, 5] supports a role for the immune system in the pathogenesis of PD.
GBA1 variants are among the most common and most important genetic risk factors of PD. Depending on the population, these variants are found in 5–20% of all PD patients [6] and are associated with a more severe, rapidly progressive phenotype [7]. Specifically, PD patients who carry GBA1 variants tend to have faster motor deterioration and cognitive decline compared to non-carriers, as well as higher frequencies of other non-motor symptoms such as hyposmia, rapid eye movement (REM)-sleep behavior disorder, hallucinations and others [7]. GBA1 encodes glucocerebrosidase (GCase), a lysosomal enzyme responsible for the degradation of glucocerebrosides and Glucosylsphingosines. The activity of GCase is reduced among individuals who carry GBA1 variants, and also in a subset of PD patients who do not have detectable GBA1 variants [8, 9], implying that GCase may also be important in some non-carriers of GBA1 variants with PD.
Gaucher disease (GD) is an autosomal recessive lysosomal storage disorder caused by biallelic GBA1 variants, with a very large spectrum of clinical presentations [10]. The association between GBA1 and PD was discovered after several clinical observations of GD patients who also developed PD [10–13]. Some patients may have very severe forms of GD with death in early childhood, whereas others may have very mild disease with almost no symptoms [14]. The variants in GBA1 can therefore be divided accordingly: “severe” variants are those associated with the severe forms of GD (type 2 and type 3), and “mild” GBA1 variants are those associated with the mild form of GD (type 1) [15]. This classification for severe and mild seems to be relevant for PD as well, since carriers of severe GBA1 variants may have higher risk for PD compared to mild variants carriers, have an earlier age at onset of PD [6], and faster cognitive decline [16, 17]. Some GBA1 variants, such as p.E326K and p.T369M, do not cause clinical GD, but are associated with PD [18, 19]. It is important to note that while GBA1 variants are common risk factors for PD, their penetrance (the probability to develop PD for someone who carries these variants) is low, between 10–30% in different studies [20–22]. In other words, most carriers of GBA1 variants will not develop PD, and the penetrance may be dependent on both genetic [23, 24] and other factors, some of which may be related to the immune system and inflammation.
One of the characteristics of GD is the presence of Gaucher cells: dysfunctional macrophages with accumulated glucocerebrosides [25]. These macrophages can accumulate in different tissues such as the spleen, liver, lung, and bone marrow. The infiltration of Gaucher cells affect the reticuloendothelial system of the respective organs and may lead to inflammation and organ dysfunction [25, 26]. Inflammation may have a key role in GD, possibly through complement activation [27] or other mechanisms, such as abnormality of cellular immunity [28], microglial activation and neuroinflammation in the neuronopathic forms of GD [29], among others.
The mechanism by which GBA1 variants increase the risk of PD is still unknown, although different hypotheses have been made, including lysosomal membrane dysfunction, the interaction between GCase substrates and α-synuclein, impaired mitochondrial function, endoplasmic reticulum stress, and impaired immune response [30]. However, the interplay between GBA1 variants, immune response, and their potential role in PD is still unclear. In this review, we summarize findings and discuss several hypotheses potentially linking GBA1, the immune system and PD.
GBA1 VARIANTS AND THE IMMUNE SYSTEM: LESSONS FROM GAUCHER’S DISEASE
GD is a rare, autosomal recessive lysosomal storage disease caused by biallelic variants in the GBA1 gene, leading to markedly reduced GCase activity and accumulation of glycosphingolipids such as glucosylceramide and glucosylsphingosine in macrophages and other cell types [29, 31] [32]. GD has a prevalence of about 1/100,000 individuals and is overrepresented in particular ethnicities, such as Ashkenazi Jews (prevalence of about 1/800 individuals) [33]. There are over 300 variants in GBA1 including point variants, deletions and insertions, and recombination variants with the nearby pseudogene [31]. GD can be classified into three types, depending on its clinical presentation: The most common form, GD type 1, has mainly peripheral signs and symptoms including splenomegaly, hepatomegaly, anemia, and acute bone pain in the pelvis and lower limbs and no apparent neuronal involvement. GD types 2 and 3, on the other hand, are characterized by severe neurological impairment, which can be acute (in type 2) or more chronic (in type 3), in addition to the typical GD symptoms seen in GD type 1 [34, 35]. However, the clinical spectrum within each type is highly variable, and it is possible to view GD more as a continuum of phenotypes rather than distinct subtypes [35].
The immune system and inflammation have an important role in the pathophysiology of all types of GD. The cells known as Gaucher cells are macrophages with extensive accumulation of glycosphingolipids, mainly glucosylceramide and glucosylsphingosine. GD patients, on top of the classical symptoms of GD, can develop a plethora of immune related symptoms including gammopathies, inflammation, and other types of immunological-related dysfunction [36]. While some of these immune-related phenomena, such as Gaucher cells, may only be relevant in GD but not in PD (where they are not typically present), it is possible that other immune-related impairments involved in GD may also have a role in PD. In this section, we will discuss several important aspects of the immune system in GD that may also be relevant in PD.
Cytokines have an important role in GD, with multiple lines of evidence from humans and different GD models showing increased pro-inflammatory cytokines [37]. For example, a study of 24 GD patients demonstrated elevated levels of different cytokines (e.g., IL-1β, IL-1 receptor antagonist, IL-6, and others) compared to control, also showing differences in cytokine levels between those who had GD type 3 compared to type 1 [38]. Whether differences in cytokine levels between severe and mild GD may explain some of the clinical differences seen in PD patients with severe versus mild GBA1 variants require additional studies. A similar study comparing 22 GD patients and 22 age- and sex-matched controls reported elevated levels of IL-6 and IL-10 in GD patients [39]. In induced macrophages stimulated with the bacterial endotoxin lipopolysaccharide (LPS), the levels of induction of the cytokine tumor necrosis factor (TNF) were significantly higher in cells induced from GD patients compared to controls [40]. Not only cytokines may be different between GD patients and healthy individuals, but also the counts of specific cell types. In a study of 18 children with GD type 1, the authors reported an increase in the number of total lymphocytes, and a specific increase in CD19+, CD3+, CD4+, and CD8+ cells [41]. In addition, another study reported a reduced number of monocytes, with decreased capacity for migration, comparing monocytes from 24 untreated GD patients and 18 controls [42]. These results and the higher levels of immunoglobulins also seen in GD patients suggest that chronic stimulation of the humoral immune system is an important characteristic of GD [43].
On top of peripheral immune response, there is accumulating evidence for neuroinflammation in GD, with a few reports on microglial activation in humans [44, 45] and in different animal models [29, 46]. Overall, the neuropathological data for GD in humans is limited, and a study reported microglial activation only in GD type 2, and its absence in GD type 3 [45]. In a neuropathological study on 14 subjects with GD, including seven with GD type 1, three with type 2 and four with type 3, no microgliosis was reported; however, analysis of microglia was not included in the study [47]. Since most of the evidence of microglial activation in GD comes from animal models, it remains to be determined if the same phenomenon is common in GD in humans, especially in GD type 1. However, in the latter study, marked astrogliosis was reported in almost all patients [47]. Astrogliosis on its own can be an important modulator of neuroinflammation [48], although this can be a reaction to neurodegeneration [49] rather than a primary part of the pathogenic process. Nevertheless, even if neuroinflammation is reactive to a neurodegenerative process, i.e., it is an outcome of neurodegeneration rather than a primary cause, it can still contribute to the neurodegenerative process as a secondary factor.
A recent, interesting imaging study used 11C-(R)-PK11195 BPND positron emission tomography (PET) scan to determine whether there is increased neuroinflammation and microglial activation in GBA1 variant carriers. Since PK11195 is a transporter protein (TSPO) ligand, and since TSPO is increased in both microglia and astrocytes during neuroinflammation, increased binding of this ligand is a marker of neuroinflammation in general, rather than only of microgliosis. In this study, the authors compared nine GBA variant carriers (five biallelic carriers and four heterozygous carriers) who do not have PD and 29 age-matched controls and reported increased neuroinflammation in brain areas that are susceptible for α-synuclein accumulation [50]. One of the interesting aspects of this observation, is that the biallelic carriers of GBA1 variants all had at least one mild variant (i.e., they would have GD type 1), and that the increased neuroinflammation was observed also in the heterozygous GBA1 variant carriers. Since at the time of the study none of the GBA1 variant carriers had PD, the results might imply that neuroinflammation or increased microglial activation is a general feature of carrying GBA1 variants. Whether or not these features have a role in developing PD in GBA1 variant carriers is yet to be determined, and since this study was small and without replication, additional studies are required to confirm this observation.
INFLAMMATION MARKERS AND CYTOKINES IN GBA1-ASSOCIATED PARKINSON’S DISEASE
In contrast to the evidence from GD patients with homozygous or compound heterozygous GBA1 variants, it is still unclear whether heterozygous GBA1 variants play a role in modulating global levels of inflammatory markers. Several studies aimed to address this question (Table 1), yet they often analyzed different cytokines with different methods and reached inconsistent results. A recent study analyzed different cytokines using two methods: ELISA was used for analyzing eight GBA1-PD patients, 28 sporadic PD patients and 28 controls, and a multiplex assay was used for analyzing eight GBA1-PD patients, 23 sporadic PD patients and 29 controls [51]. In the ELISA analysis, levels of IL-1β and TNFα were both significantly higher among the GBA1-PD group, and IFNγ was lower in GBA1-PD compared to sporadic PD. However, in the multiplex assay, only the results for IL-1β were replicated. TNFα was not different between the two groups of patients, and it was lower compared to controls. IFNγ, which in the ELISA analysis was lower in GBA1-PD compared to sporadic PD, was much higher in the multiplex assay in GBA1-PD compared to both sporadic PD patients and controls. The multiplex assay also analyzed cytokines that were not measured using ELISA, and suggested that the levels of IL-2 and MCP-1 (monocyte chemoattractant protein-1, also known as chemokine (c-c motif) ligand 2, CCL2), a cytokine that regulates migration and infiltration of monocytes [52] are increased in GBA1-PD.
Summary of studies analyzing different inflammation markers and cytokines in GBA1-PD
ELISA, enzyme-linked immunosorbent assay; PD, Parkinson’s disease; IL, interleukin; TNF, tumor necrosis factor; IFN, interferon; MCP-1, monocyte chemoattractant protein-1, also called CCL2; MIP1α, macrophage inflammatory protein 1α, also called CCL3; PARC, Pulmonary and activation-regulated chemokine, also called CCL18.
A previous report comparing 20 GBA1-PD patients (17 heterozygous and three homozygous) and 242 sporadic PD patients also reported increased plasma levels of MCP-1 in GBA1-PD [53]. The same study also reported increased levels of IL-8, MIP1α (macrophage inflammatory protein 1α, also known as chemokine (c-c motif) ligand 3, CCL3) and several other cytokines in GBA1-PD. However, in a replication cohort of 19 GBA1-PD patients (17 heterozygous and two homozygous) and 41 sporadic PD patients, only IL-8 showed increased levels in GBA1-PD [53]. A recent study comparing 77 GBA1-PD patients with 31 idiopathic PD patients, however, did not identify association between GBA1-PD and any of the tested cytokines, including TNF-α, IL-1, IL-2, IL-4, IL-6, IL-8, IL-10, and INF-γ [54]. A different study evaluated the levels of established GD-associated cytokines in biallelic carriers of GBA1 variants with PD (n = 6, four of which were also diagnosed with GD), heterozygous carriers of GBA1 variants with PD (n = 129), individuals with GBA1 variants without PD (n = 83) and controls without GBA1 variants (n = 77) [55]. The analysis demonstrated levels of ferritin, CCL18, and MIP1α were only elevated in the group of biallelic GBA1 variants with PD. Of note, some of the chemokines reported in the studies above to potentially be involved in GBA1-PD, including IL-8, MIP1α, TNF-α, and MIP1α, have been suggested to predict the progression of LRRK2-associated PD, another common genetic form of PD [56]. Their putative role in GBA1-PD progression has not been studied yet, and direct comparisons between GBA1-PD and LRRK2-PD will be of much interest and importance.
There are many differences between the studies mentioned above that can explain the contradicting results, including differences in sample size, methods used for analysis, ethnicity, environmental factors and individual genetic factors that can affect the levels of cytokines. A recent study aimed to standardize different assays in immune cells, including measurement of cytokines, suggested that performing stimulation-dependent analysis will yield more consistent results across different centers [57]. A coordinated, large effort of multiple centers that will use standardized methods and analysis, accounting for as many confounders as possible, is required to better understand whether specific cytokines have a role or can serve as biomarkers in GBA1-PD. Furthermore, not all the aforementioned studies considered the different types of GBA1 variants, and therefore included all variant carriers as a single group. It will be important in future studies to separately analyze the effects of different types of GBA1 variants (severe, mild, risk factors, specific variants) and to detail exactly which variants were analyzed, in order to fully understand how they might specifically contribute to PD through immune response.
GLYCOSPHINGOLIPIDS AND NEUROINFLAMMATION
Several genes in the lysosomal sphingolipid metabolism pathway have been suggested to be involved in PD. These include, GBA1, SMPD1, ASAH1 [59], GALC [5], ARSA [60], and GLA [61], and their main substrates, products, related lysosomal storge disorders and suggested involvement in PD are detailed in Table 2. All these genes work closely together in the sphingolipid metabolism pathway in the lysosome (Fig. 1), suggesting that dysregulation of the flux in this pathway may be important in PD pathogenesis. Other genes that regulate GBA1 have also been implicated in α-synucleinopathies, including PSAP [62, 63], SCARB2 [64, 65], and CTSB [23].
Genes for known lysosomal storage diseases and their respective genetic associations to Parkinson’s disease
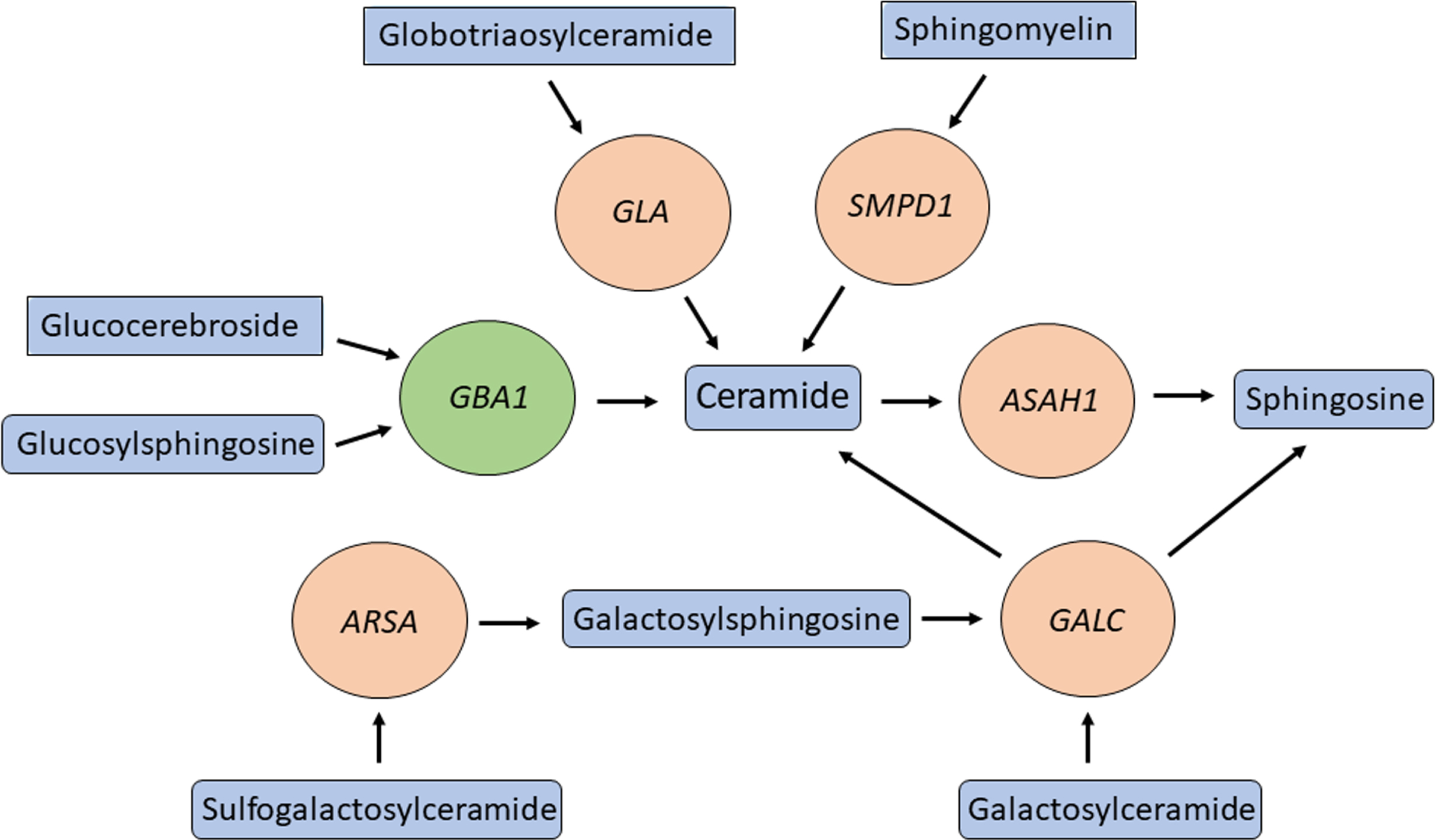
Multiple genes involved in ceramide metabolism and in PD. GBA1 (green circle) is one of many genes (orange circles) encoding for enzymes in the ceramide metabolism pathway that are both responsible for a lysosomal storage disease and modifying the risk of PD. Important substrates and products of these enzymes are in blue rectangles.
At the same time, the sphingolipids that are substrates and/or products of this pathway have important immune-related and inflammatory roles. This topic has been thoroughly reviewed in relation to PD [66], therefore here we provide some important details and takes. Glucocerebrosides (or glucosylceramides) are the main substrates of all glycosphingolipids in the brain. In general, glycosphingolipids regulate multiple processes in the brain, including immune response and neuroinflammation. One mechanism linking glycosphingolipids and neuroinflammation is activation of inflammasomes— a complex of proteins activating caspase-1, which is responsible for the maturation of the proinflammatory cytokines IL1-beta and IL18. Autophagy and the inflammasome regulate each other [67], and efficient autophagy is critically regulated by the composition of membranes, which is determined by the levels of the different sphingolipids [68–70]. Whether this link between GCase and other enzymes in the sphingolipids metabolism pathway, autophagy and inflammasome activation has a role in PD remains hypothetical and requires additional studies.
Another potential link between the lysosomal membrane composition and the immune system in PD is through antigen presentation. There is a clear association between the HLA genomic region and specific HLA types and PD [2, 3], and the process of antigen processing and presentation is regulated by autophagy and the lysosome [71]. It is therefore possible that GBA1 variants, as well as variants in other genes in this pathway, affect the lysosomal membrane composition, which affects autophagy and antigen presentation. This hypothesis, as does the former one, require additional studies in humans and different models. Other mechanisms by which glycosphingolipids may affect neuroinflammation include their effects on calcium homeostasis, blood-brain-barrier permeability and autoantibodies production [66].
Lastly, there is a large body of evidence on the interaction between α-synuclein and membranes, which has been extensively reviewed before [72]. Since GBA1 variants affect membrane composition, it is possible that they influence the interaction between these membranes and α-synuclein. These hypothesized effects should be further studied in the context of GBA1-PD in different PD-relevant models and cell types.
GBA1, α-SYNUCLEIN, AND THE IMMUNE SYSTEM
Unlike another common genetic form of PD, LRRK2-associated neurodegeneration, GBA1-associated PD is almost exclusively characterized by α-synuclein neuropathology [73]. Numerous studies in multiple human and animal show that reduced GCase activity, whether due to variants, chemical inhibition, knockdown or knockout, leads to α-synuclein accumulation [74–76]. Furthermore, it was suggested that GCase, its substrate glucocerebrosidase and α-synuclein directly interact as part of the pathogenic process in GBA1-associated PD [77, 78].
Despite decades of research, it is still unclear what the normal, physiological role of α-synuclein is. It is possible that one of its potential roles is as a part of the immune system [79], and whether this hypothesis is true or not, years of evidence clearly show that α-synuclein can trigger the immune system, and that it may have a role in the pathogenic process of PD [80]. Specifically, aggregated α-synuclein can activate microglia in vitro and in vivo [81], which in turn may contribute to the pathogenic process of PD. This activation may occur through binding to the Toll-like receptor 2 (TLR2), as mice lacking TLR2 or treated with TLR2 blocking antibody did not show microglial activation [82]. Other lines of evidence show that α-synuclein can also trigger the activation of T cells. Certain epitopes of α-synuclein may act as antigens and stimulate helper and cytotoxic T cell responses in PD patients [83], perhaps due to their homology to some viruses such as the herpes simplex virus 1 (HSV1) [84]. It is still unclear whether this activation is part of the normal function of α-synuclein in attempt to activate the immune system as a defence mechanism, or a pathogenic process that in some individuals may contribute to the development of PD, or a combination of both scenarios. While this is not a unique feature of GBA1-associated PD and may occur in other forms of PD, it represents another path by which the immune system participates in this specific PD subtype.
ADDITIONAL EVIDENCE POTENTIALLY LINKING GBA1 AND THE IMMUNE SYSTEM WITH PD
The complement system plays an important role in assisting immune cells with clearing infections and modulating immunity. Dysregulation of the complement system leads to various effects from autoimmune disorders to failures in host immunity. Furthermore, the complement system has been implicated in normal central nervous system development and in different neurological disorders, including PD [85, 86]. Activated complement components have been reported in neuropathological studies of brains of PD patients and observed within Lewy bodies. Changes in complement levels in blood of PD patients has also been reported, and in vitro studies suggest that α-synuclein may have a role in their activation [87].
A few studies pointed towards a potential involvement of the complement system in GBA1-mediated dysregulation of the immune system. GBA1 mutations may result in activation of the complement pathway via increased production of C5a and activation of the C5aR1 receptor which in turn results in the production of pro-inflammatory cytokines. Compared to wildtype, mice with heteroallelic mutations (p.Asp409Val/knockout) in GBA1 had a marked increase in C5a, and increased levels of inflammatory cells and cytokines including IFNγ, TNF, IL-1β, IL-6, and IL-17A/F [27]. Of note, this specific mutation has been reported only once in GD, and never in PD, and its severity is unknown. Similarly, GD-derived macrophages (using induced pluripotent stem cells) showed marked production of TNF in response to C5aR1 activation in the presence of recombinant C5a [88]. Both of these studies point towards a possible role for the complement system in potentiating an inflammatory environment due to GBA1 variants.
Another potential link between GBA1 variants and immune response was demonstrated in mice, where GCase inhibition with conduritol-β-epoxide (CBE) led to impaired response of the nuclear factor erythroid 2-related factor (Nfe2l2), a key transcription factor involved in multiple processes, including neuroinflammation. Using neuronal-microglial co-cultures derived from the same mouse models revealed that the inhibition of the response was mediated by inhibition of GCase activity in microglia and not in neurons themselves. Furthermore, GCase inhibition in microglia was sufficient and necessary for ablation of the Nfe2l2 in neurons [89]. Another study showed that mice treated with continuous administration of GCase inhibitor resulted in prolonged activation of microglia, the complement pathway, and increased expression of autophagy-related proteins in the substantia nigra after 28 days of treatment compared to mice treated with vehicle [90]. In long-lived transgenic mouse models with homozygous p.L444P (associated with GD type 3) or p.R463C (associated with GD type 1) GBA1 variants had increased activation of astrocytes and accumulation of alpha-synuclein in the nigrostriatal pathway. Furthermore, in the same models, subchronic administration of GCase inhibitor also resulted in activation of microglia and astrocytes in the cortex, hippocampus, substantia nigra and striatum, as well as marked increases in alpha-synuclein accumulation in the substantia nigra [91].
One interesting aspect of the potential involvement of GBA1 in immune response and inflammation, is that its substrates and products may also have a role. For example, in monocytes, inhibition of ceramide kinase (CERK) leads to reduction in expression of inflammatory markers CD11c and HLA-DR by monocytes in response to TNF-α stimulation [92]. Glucosylceramide, one of the main substrates of GCase, can activate myeloid cells and increase the levels of inflammatory cytokines [93]. Conversely, reduction of glucosylceramide can reduce LPS-induced inflammatory response in human macrophages and in mice [94]. Other studies have shown that the activity of the GCase itself might be specifically reduced in monocytes, even in non-carriers of GBA1 variants, and that this reduced activity may correlate with the motor severity of PD [95, 96]. These are just a few examples, but multiple other studies link glucosylceramide and other glycosphingolipids to immune homeostasis [97], providing an additional potential link between GBA1 and inflammation. Due to contradicting results it is still unclear whether in PD with heterozygous GBA1 variant there is an elevation of glucosylceramide, glucosylsphingosine, or other GCase substrates [98], suggesting that other mechanisms may be involved.
CONCLUDING REMARKS
The goal of this review was to provide an overview of the current evidence linking GBA1, the immune system and PD. While there is some suggestive evidence that the immune system and/or inflammation may be involved in GBA1-associated PD (Fig. 2), it is far from being conclusive. The differences between the different studies which examined cytokine levels in GBA1-PD patients require additional studies that at the very least should be: a) larger, b) use the same methodology, and c) take into account other confounding factors like genetics, co-morbidities and different exposures. Even if there are differences in cytokine levels, showing that they have a role in the pathogenic process of GBA1-associated PD will require additional studies. Similar questions remain regarding all other evidence that we discussed in this review, and their potential role in GBA1-associated PD. Since there are many drugs that can modulate the immune system and inflammation, it will be important to address these questions.
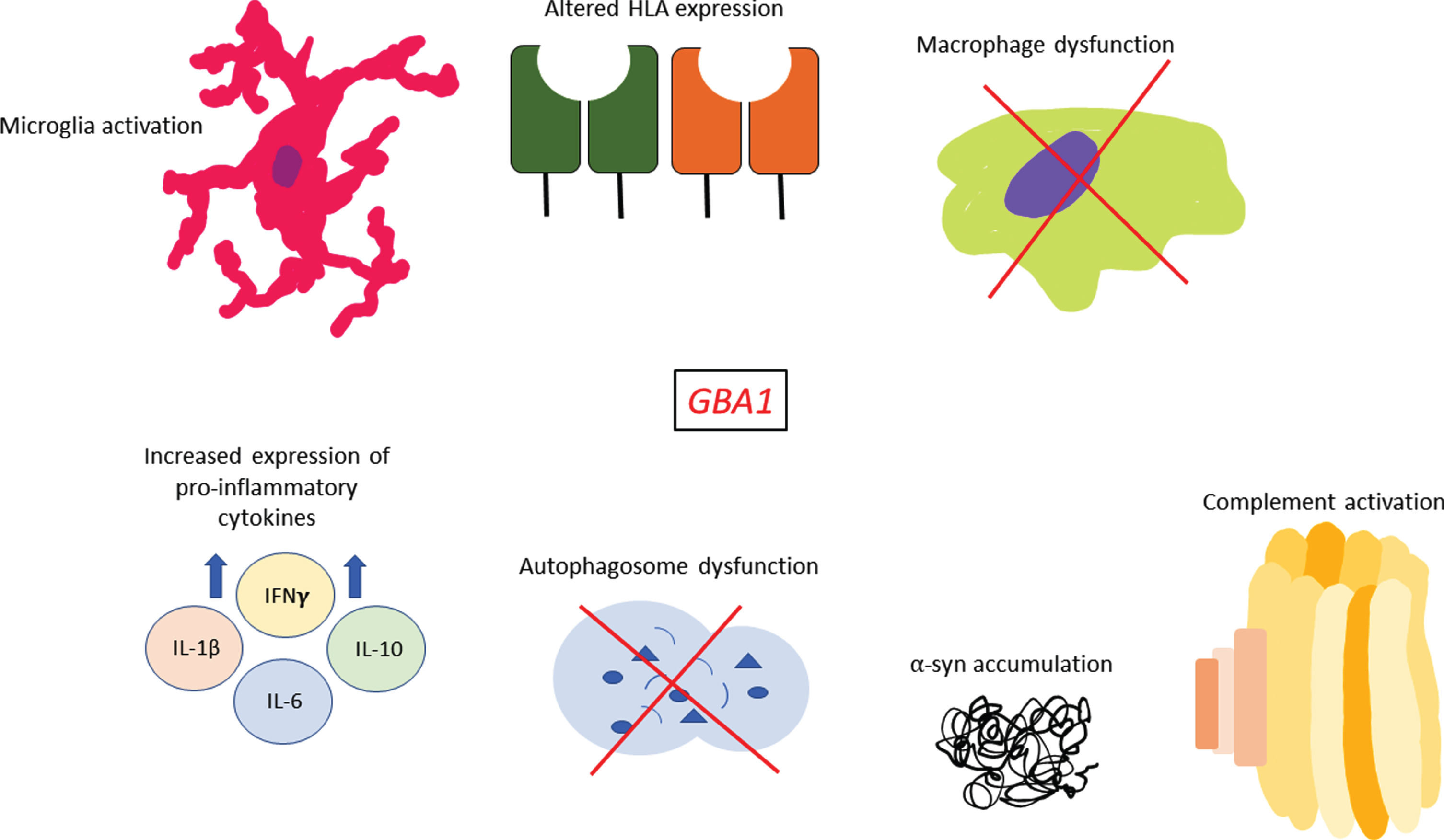
Sequelae of immune events associated with GBA1 variants. GBA1 variants have been shown to affect multiple facets of the immune system such as increased pro-inflammatory cytokine expression, complement activation, potentially altered HLA expression, microglia activation and macrophage dysfunction. In addition, many studies have shown that GBA1 variants are associated with greater α-synuclein accumulation within cells and marked autophagosome dysfunction which may also have immune-related effects in PD.
Footnotes
ACKNOWLEDGMENTS
This work was financially supported by the Michael J. Fox Foundation, the Canadian Consortium on Neurodegeneration in Aging (CCNA), and the Canada First Research Excellence Fund (CFREF) awarded to McGill University for the Healthy Brains for Healthy Lives (HBHL) program.
CONFLICT OF INTEREST
Z.G.O. is supported by the Fonds de recherche du Quebec Sante (FRQS) Chercheurs-boursiers award. He received consultancy fees from Ono Therapeutics, Handl Therapeutics, UCB, Neuron23, Lysosomal Therapeutics Inc., Bial Biotech Inc., Deerfield, Lighthouse, Gudiepoint, and Idorsia.