
Abstract
Background
The adaptive immune response has a role in Parkinson's disease (PD). Patients with LRRK2 or GBA1 mutations often exhibit distinct clinical characteristics.
Objective
To evaluate the involvement of adaptive immune response genes in three PD groups: GBA1-PD, LRRK2-PD, and non-carrier (NC)-PD.
Methods
Differentially expressed genes (DEGs) associated with PD were identified using four datasets. Of them, adaptive immune response genes were evaluated using whole-genome-sequencing of 201 unrelated Ashkenazi-Jewish (AJ) PD patients. Potential pathogenic variants were identified, and P2RX7 variants were assessed in 1200 AJ-PD patients. Burden analysis of rare variants (allele frequencies (AF) < 0.01) on disease risk, and association analyses of common variants (AF ≥ 0.01) with disease risk and age-at-onset (AAO) were conducted. AFs were compared to AJ-non-neuro cases reported in gnomAD. Variants associated with PD were further examined in an independent AJ cohort from AMP-PD.
Results
Of the four adaptive immune DEGs identified, CD8B2, P2RX7, IL27RA, and ZC3H12A, three common variants in P2RX7 were statistically significant: Tyr155His was associated with NC-PD (allelic OR = 1.15, p = 0.015) ; Arg276His was associated with LRRK2-PD (allelic OR = 2.10, p = 0.037), while Glu496Ala was associated with earlier AAO in LRRK2-PD (p = 0.014). Burden analysis showed no significant effect on PD-risk. In the AMP-PD cohort, odds ratios of the two risk variants were similar to the primary cohort, but did not reach significance, probably due to small control sample size (n = 263).
Conclusions
Common variants within P2RX7 are likely associated with PD-risk and earlier AAO. These findings further suggest P2RX7's involvement in PD and its potential interplay with LRRK2.
Plain language summary
Plain Language Summary: Among many causes, evidence suggest that the immune system plays a role in the development of Parkinson's disease (PD). This study focused on understanding how certain immune-related genes might be involved in PD risk, including people who carry already known genetic risk mutations in LRRK2 and GBA1 genes. Four genes related to the adaptive immune response, CD8B2, P2RX7, IL27RA, and ZC3H12A, demonstrated different expression in the substantia nigra (a primary brain region of neuronal loss in PD) between PD patients and healthy individuals. We focused on P2RX7 (Purinergic Receptor P2X 7) gene and examined the potential enrichment of its genetic variations in a large group of 1200 PD patients of Ashkenazi-Jewish origin, compared to healthy individuals, which may suggest genetic involvement in the disease. Three common DNA variations in this gene were found to be associated with PD. Two of them were linked to a higher risk of PD, and another one was linked to an earlier disease onset in patients who carry a LRRK2 mutation. P2RX7 activation has been linked to inflammation. Our results suggest that this gene could play a role in PD development, which may lead to new approaches for treatment and prevention.
Introduction
Parkinson's disease (PD) is a progressive neurodegenerative disorder characterized by both motor and non-motor symptoms. While the precise etiology of PD is poorly understood, it is known to involve a complex genetic contribution. Among the most common genes associated with PD are Leucine-Rich Repeat Kinase 2 (LRRK2) and Glucosylceramidase beta 1 (GBA1), which contain both common and rare variants linked to PD risk. 1
Variants within the LRRK2 gene have been identified as associated with an increased risk of PD, with its most common variant, the G2019S variant (rs34637584), present in approximately 1% of sporadic PD cases and 4% of familial cases. 2 GBA1 mutations lead to Gaucher Disease when present in homozygous or compound heterozygous states and increase the risk of developing PD in the heterozygous state. 3 Patients with LRRK2 mutations often display distinct clinical and pathological features compared to those with GBA1 mutations, particularly in terms of disease progression, cognitive deficits, and Lewy body pathology. 4
Although conflicting evidence exists regarding whether the immune response differs among PD patients who carry variants in LRRK2 or GBA1,5–7 it is well-established that activation of the immune system is involved in the pathophysiology of PD. Specifically, several studies have suggested a role for adaptive immunity in PD.1,8–14
While the innate immune response, which involves cells such as monocytes, macrophages, neutrophils, and natural killer cells, acts as the body's first defense mechanism, the adaptive immune response involves two main classes of lymphocytes: T cells and B cells. These lymphocytes mount specific responses against foreign antigens and develop immunologic memory. Recent studies have shed light on the dysregulation of the adaptive immune response observed in PD. One study demonstrated a proinflammatory profile in early-stage PD patients, and reduced levels of naive CD4 + and CD8+ T cells compared to controls. 12 Another study, using induced pluripotent stem cells from midbrain neurons of PD patients, found an elevated number of T-lymphocytes in these cells, and higher levels of T-helper 17 cells in the blood of PD patients. 9 Furthermore, genetic studies have revealed an association between polymorphisms in human leukocyte antigen, a gene that plays a role in activating the adaptive immune response, and sporadic late-onset PD.1,13,14 In a large-scale pathway analysis using whole-genome sequencing (WGS) data, the burden of rare loss-of-function (LoF) variants was found to be associated with PD through the adaptive immune system pathway. 8 Taken together, these findings support the involvement of adaptive immunity in the pathogenesis of PD.
In this study, we aimed to interrogate the involvement of the adaptive immune response genes in PD, within three subgroups: GBA1 carriers, LRRK2-G2019S carriers, and non-carriers (NC).
Methods
Gene expression dataset
The Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) was used to identify datasets from studies investigating changes in gene expression in the substantia nigra (SN) of PD patients and controls. The focus was on the SN as it is the primary region of neuronal loss and is involved in PD pathology. To identify differentially expressed genes (DEGs), suitable datasets were inquired using several keywords, including “Parkinson's Disease”, “Substantia nigra”, “Homo sapiens”, and “Expression profiling by high throughput sequencing”. Four datasets, generated via high-throughput sequencing of the SN, from both PD patients and controls, were selected: GSE114517 (included 17 PD patients and 12 controls), 15 GSE136666 (included 5 PD patients and 5 controls), 16 GSE133101 (included 15 PD patients and 10 controls), 17 and GSE168496 (included 8 PD patients and 8 controls) 18 (Supplemental Table 1, Supplemental Figure 1).
Adaptive immune genes target list
Using the Molecular Signature Database (MSigDB) (https://www.gsea-msigdb.org/gsea/msigdb), genes included in the ‘Adaptive Immune Response’ category (GO:0002250) were retrieved.
Whole genome sequencing (WGS) and quality filters
WGS was performed on 201 unrelated PD patients of Ashkenazi Jewish ancestry (AJ) as previously described.19,20 Briefly, sequencing was performed using DNBseq technology at BGI, China. Paired-end reads with a length of 100 bp and a depth coverage of 30X were utilized. Variant call format files were generated for each sample by aligning to the human reference genome GRCh38/hg38 build (hg38) using the BWA tool. 21 Subsequently, the Genome Analysis Toolkit (GATK) was utilized for variant calling on the alignment data of each sample, as previously described. 22
Variants were extracted from the selected adaptive immune DEGs (n = 4), with an additional 1Kb flanking each gene (±1Kb), using SNP & Variation Suite V.8.9.0 (Golden Helix, Inc), and those with a read depth below 10 and genotype quality under 30 were excluded. After additional filtration steps, the remaining variants were those with more than one carrier and combined annotation-dependent depletion (CADD, version 1.6 23 ) Phred scores equal to or greater than 20 (Supplemental Figure 1). The effect of variants on gene expression (expression quantitative trait loci) was evaluated in GTEx Portal database (https: //gtexportal.org/, accessed on 1 March 2023).
Genotyping
Our PD cohort includes 1200 unrelated patients of full AJ origin (self-reported, 171 of them were included in the cohort of 201WGS PDs), recruited consecutively between 2005 and 2016 (with an average age of motor symptoms onset of 60.56 ± 10.96). All patients underwent examination at the laboratory for Early Markers of Neurodegeneration (LEMON) at the Tel Aviv Sourasky Medical Center, Israel, and diagnosis was determined by movement disorders specialists.24,25 Among these, 235 (19.6%) were carriers of GBA1 mutations (GBA1-PD) (severe: p.L444P, c.84insG, IVS2 + 1G > A, p.V394L, mild: p.R496H, p.N370S, 370Rec, and the risk alleles: p.E326 K and p.T369 M, p.R44C); 145 (12.1%) were carriers of LRRK2-G2019S (LRRK2-PD); eight (0.6%) were carriers of the SMPD1-L302P mutation; 25 (2.1%) carried mutations in more than one gene (24 GBA1 and LRRK2 G2019S carriers, and one GBA1 and SMPD1 L302P carrier); and 787 (65.6%) did not carry any of these GBA1, LRRK2, or SMPD1 mutations (non-carriers, NC-PD) (Supplemental Table 2).
To confirm AJ ancestry and rule out relatedness between the patients, principal component analysis (PCA) and identity-by-descent (IBD) analysis were conducted on a subset of 591 patients from the 1200 PD patients cohort, using Affymetrix Genome-Wide Human SNP Array 6.0 data (the Tel Aviv PD SNP6.0 array data as previously described 26 ). PCA and IBD analyses were conducted on the WGS cohort (n = 201) as well.
The complete cohort of 1200 PD patients underwent genotyping for the eight P2RX7 variants (6 missense and 2 LoF donor-site variants, Supplemental Table 3) using the StepOnePlus system (Applied Biosystems). Samples with ambiguous calls for rs208294 were further sequenced using Sanger sequencing (Forward- 5’-CTTGTGTTCGTTGTGGTTAGGAT-3’ Reverse- 5’-CTGCAAAGTCCTGGGTCCTAC-3’, Supplemental Table 3).
Secondary cohort: Accelerating Medicine Partnership Parkinson’s Disease (AMP-PD)
The Accelerating Medicines Partnership Parkinson's disease (AMP-PD) cohort was used as a secondary confirmation cohort. Samples were sourced from the Parkinson's Progression Markers Initiative (PPMI), Harvard Biomarkers Study (HBS), Parkinson's Disease Biomarkers Program (PDBP), and the BioFIND study. We looked at unrelated AJ samples and excluded those who were analyzed in our primary cohort. A total of 584 individuals were included, 321 PD patients and 263 controls (Supplemental Table 2). Within this cohort, 162 were carriers of one or more GBA1 mutations (those mutations described above; 74 PD patients and 88 controls), 224 carried the LRRK2 G2019S mutation (125 PD patients and 99 controls), 20 were dual carriers (GBA1 and LRRK2 G2019S; 10 PD patients and 10 controls) and 178 did not carry any of these GBA1, LRRK2, or SMPD1 mutations (112 PD patients and 66 controls). In this cohort, AJ ancestry was already determined 27 and IBD was performed on WGS data using PLINK 1.9. 28
In silico characterization of identified risk variants
To further characterize the identified variants associated with PD and predict their pathogenicity, various in silico tools were used. DUET was used to predict changes in protein stability, 29 ConSurf to assess the evolutionary conservation of amino acid positions, 30 and Aminode to detect evolutionarily constrained regions. 31
Statistical analysis
The association of common variants in P2RX7 was assessed by comparing PD patients from our cohort (1200 AJ-PD cohort) to the gnomAD dataset of AJ-non-neuro (v2.1.1), which served as the control cohort and will hereafter be referred to as gnomAD-AJ-nn-controls. This control dataset comprises samples from individuals of AJ ancestry who were not ascertained for neurological conditions in case/control studies. 32 Common variants were defined as those with an allele frequency (AF) ≥ 0.01 in the gnomAD-AJ-nn-controls. Odds ratios (ORs) and 95% confidence intervals (CIs) were calculated using ‘MedCalc’ (https://www.medcalc.org).
Burden analysis for rare variants, defined as those with an allele frequency (AF) < 0.01 in the gnomAD-AJ-nn-controls cohort, was conducted by comparing the number of individuals carrying rare alleles in our PD cohort to those predicted to carry these variants in gnomAD-AJ-nn-controls, calculated using allele frequencies, and under the assumption that none will carry more than one allele (a two-tailed Fisher's exact test, ‘GraphPad’ https://www.graphpad.com/quickcalcs/contingency1/).
The effect of common variants on age-at-motor-symptoms onset (AAO) was investigated using SPSS Statistics software version 25 (t-test in GBA1-PD and LRRK2-PD, and Mann–Whitney U test in NC-PD). We have previously shown that the AAO of GBA1-PD was influenced by mutation dosage and the severity of GBA1 mutations (severe or mild). 25 To prevent any confounding effects, we excluded carriers of severe GBA1 mutations, compound heterozygotes, and N370S homozygotes from the AAO analysis (a total of 67 patients were excluded).
Results
Adaptive immune genes with differential expression in the substantia nigra of PD patients
One hundred and forty-two genes were differentially expressed in the SN between PD patients and controls in the four GEO datasets. More specifically, 11 DEGs (8 downregulated and 3 upregulated) in GSE114517, 112 DEGs (62 downregulated and 50 upregulated) in GSE136666, 10 DEGs (2 downregulated and 8 upregulated) in GSE133101, and 24 DEGs (17 downregulated and 7 upregulated) in GSE168496 were identified (Supplemental Figure 1). Out of the 142 DEGs, only four were classified as part of the adaptive immune response gene set (GO:0002250, n = 745). One gene, CD8B2, was downregulated in PD patients, while three genes, P2RX7, IL27RA, and ZC3H12A, were upregulated.
Variant extraction
A total of 1521 variants were identified in the WGS of 201 PD patients within the four adaptive immune genes, CD8B2, P2RX7, IL27RA, and ZC3H12A. After filtering (see Methods), 10 variants remained. One was located 1Kb upstream of IL27RA but was annotated to PALM3, which is not an adaptive immune response gene, and does not affect IL27RA RNA levels (GTEx), and therefore was excluded from further evaluation. The rest of the nine variants included one missense variant within CD8B2 and eight variants within P2RX7 (six missense and two intronic-donor-site, Supplemental Table 3). As increasing evidence suggests P2RX7 is a potential target in neurodegenerative diseases,33,34 we further explored and assessed the genetic contribution of this gene to PD risk and AAO. Among the eight P2RX7 variants, three were common (with AF ≥ 0.01), while five were rare, as reported in gnomAD-AJ-nn-controls (Supplemental Table 3). All eight variants were genotyped in a larger cohort of 1200 PD patients. We conducted a risk analysis for the three common variants and a burden test for the five rare variants.
P2RX7 variants are associated with PD risk
Risk analysis of common variants. To evaluate the contribution of the three common variants within P2RX7 (AF ≥ 0.01), we performed risk analysis for each variant, using a larger cohort of 1200 PD patients (Table 1). rs208294 (P2RX7 Tyr155His) was significantly associated with PD risk among NC-PD (allelic OR = 1.147, CI = 1.03–1.28, p = 0.015; Table 1) this variant was also significant under a dominant model (genotypic OR = 1.123, CI = 1.01–1.50, p = 0.036). Additionally, this variant was significantly associated with PD risk in the complete cohort of 1200 PD patients (allelic OR = 1.145, CI = 1.04–1.26, p = 0.005; Table 1) and stayed significant under a dominant model (genotypic OR = 1.122, CI = 1.04–1.44, p = 0.016).
Risk analysis results of P2RX7 variants.
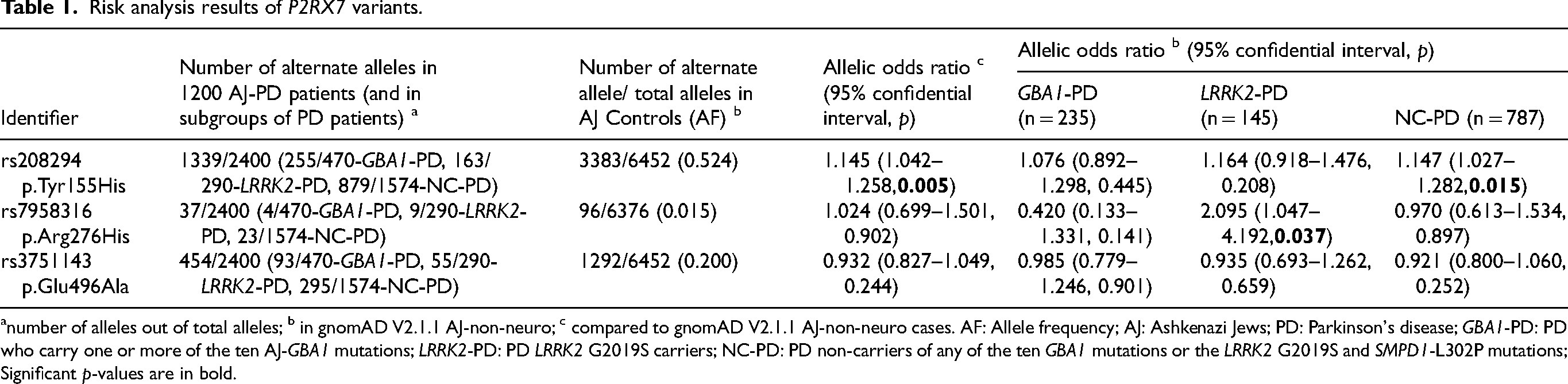
number of alleles out of total alleles; b in gnomAD V2.1.1 AJ-non-neuro; c compared to gnomAD V2.1.1 AJ-non-neuro cases. AF: Allele frequency; AJ: Ashkenazi Jews; PD: Parkinson's disease; GBA1-PD: PD who carry one or more of the ten AJ-GBA1 mutations; LRRK2-PD: PD LRRK2 G2019S carriers; NC-PD: PD non-carriers of any of the ten GBA1 mutations or the LRRK2 G2019S and SMPD1-L302P mutations; Significant p-values are in bold.
rs7958316 (P2RX7 Arg276His) was significantly associated with LRRK2-PD (allelic OR = 2.095, CI = 1.047–4.192, p = 0.037; compared to gnomAD V2.1.1 AJ-non-neuro, Table 1). The dual carrier rate of LRRK2-PD patients that also carried P2RX7 Arg276His variant was 6.2% (9/145), which is significantly higher than the 3% LRRK2 G2019S/P2RX7 Arg276His dual carrier rate predicted by gnomAD-AJ-nn-controls allele frequencies (that do not carry GBA1 mutations or SMPD1 L302P; allelic OR = 2.140, CI = 1.035–4.422, p = 0.040 when simulating on 100,000 AJ). The variant rs3751143 (P2RX7 Glu496Ala) exhibited no association with either PD risk or protection in any of the subgroups or in the entire cohort.
P2RX7 rare variants burden analysis. The five P2RX7 rare variants were genotyped in our 1200 AJ-PD cohort and the total numbers of carriers for any of the five variants were compared to the predicted total number of carriers in gnomAD-AJ-controls. There was no significant difference in P2RX7 burden between PD patients and controls (p > 0.05).
P2RX7 Glu496Ala is associated with early AAO in LRRK2-PD
P2RX7 Glu496Ala carriers in the LRRK2-PD subgroup exhibited a significantly earlier AAO (55.56 ± 10.63) compared to LRRK2-PD who did not carry P2RX7 Glu496Ala (60.04 ± 10.14, p = 0.014, Table 2). Given the relatively high AF of P2RX7 Tyr155His, we also conducted an AAO analysis of P2RX7 Glu496Ala among carriers of both LRRK2 G2019S and P2RX7 Tyr155His (N = 118) to rule out confounding. The significance remained (p = 0.012), indicating that the effect on AAO is driven by P2RX7 Glu496Ala. Additionally, neither P2RX7 Tyr155His nor P2RX7 Arg276His showed a significant effect on AAO in PD subgroups (Table 2).
Stratified age-at-motor-symptoms onset (AAO) analysis of P2RX7 variants.
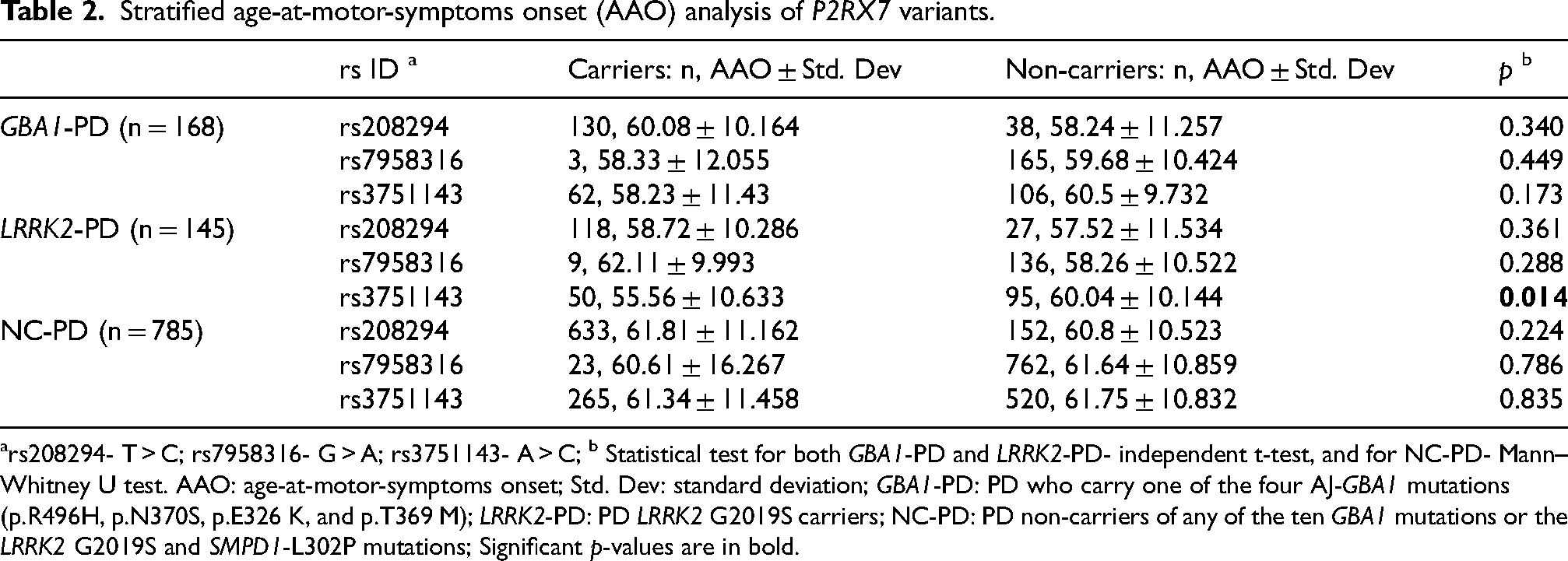
rs208294- T > C; rs7958316- G > A; rs3751143- A > C; b Statistical test for both GBA1-PD and LRRK2-PD- independent t-test, and for NC-PD- Mann–Whitney U test. AAO: age-at-motor-symptoms onset; Std. Dev: standard deviation; GBA1-PD: PD who carry one of the four AJ-GBA1 mutations (p.R496H, p.N370S, p.E326 K, and p.T369 M); LRRK2-PD: PD LRRK2 G2019S carriers; NC-PD: PD non-carriers of any of the ten GBA1 mutations or the LRRK2 G2019S and SMPD1-L302P mutations; Significant p-values are in bold.
P2RX7 variants in a secondary PD cohort (AMP-PD)
In a secondary independent cohort (AMP-PD), AFs of both P2RX7 Tyr155His and Arg276His were similar to those in our PD cohort (for Tyr155His in NC-PD, 0.562 compared to 0.558, p = 0.943 and for Arg276His in LRRK2-PD, 0.032 compared to 0.031, p = 1.000, respectively). Additionally, the AFs of these variants were higher in PD patients compared to controls (for Tyr155His in NC, OR was 1.04, and for Arg276His in LRRK2 G2019S carriers, OR was 3.24). However, these results did not reach significance, probably due to the relatively small cohort of AJ controls (n = 263) in AMP-PD (Supplemental Table 2).
In silico variants characterization
The three common variants associated with PD or earlier AAO, P2RX7 Tyr155His, Arg276His, and Glu496Ala, were predicted to decrease protein stability (Table 3). Additionally, both Arg276His and Glu496Ala were found to be relatively conserved and located in an evolutionarily constrained region (Table 3).
In silico characterization of common P2RX7 variants associated with PD risk or age-at-motor-symptoms onset.

Scale is 1–9 (9 being the most conserved); CADD: Combined Annotation Dependent Depletion; DDG: the variation in Gibbs Free Energy.
Discussion
We identified four DEGs in PD that are related to the adaptive immune response (using public database GEO). We further investigated the genetic involvement of P2RX7 in PD and identified three common variants associated with PD risk or AAO. However, we found no significant difference in the burden of rare variants between PD patients and controls.
P2RX7 functions as a ligand-gated cation channel, activated by high concentrations of ATP, allowing the passage of cations such as Na+, K+, and Ca2+. 35 Evidence suggests that P2RX7 is associated with PD. For example, in a murine cellular model of PD, it was demonstrated that α-synuclein interacts with microglial P2RX7, resulting in increased ROS production and enhanced neuronal loss. 36 These effects were inhibited after P2RX7 knockdown. 36 Other in vitro and in vivo studies in rat models for PD indicated that treatment with the P2RX7 antagonist, Brilliant Blue G (BBG), attenuated neurotoxicity and the loss of dopaminergic neurons.37–39 Moreover, in a genetic study on the Han Chinese population, it was suggested that a P2RX7 Glu496Ala is associated with an increased risk for PD. 40
In the central nervous system, P2RX7 is primarily expressed in microglia, astrocytes, and oligodendrocytes. In microglial cells, stimulation of P2RX7 triggers the activation of the NLRP3 inflammasome, resulting in the release of pro-inflammatory cytokines such as interleukin-18 (IL-18) and interleukin-1β (IL-1β). 34 Multiple studies have indicated that increased activation of the NLRP3 inflammasome and elevated secretion of IL-18 and IL-1β are associated with PD.41,42 Additionally, P2RX7 stimulation induces the secretion of tumor necrosis factor-α (TNF-α) and the generation of reactive oxygen species (ROS, a known key modulator in PD pathogenesis) leading to neuroinflammation and cell death. 34 Altogether, these findings support a potential role for P2RX7 in PD pathophysiology and suggest it as a potential therapeutic target in PD.
In our study, we showed that the P2RX7 p.Tyr155His (T > C) variant is associated with PD risk in both stratified (NC-PD patients) and unstratified (the entire cohort of PD patients) analyses. Notably, the C allele (resulting in histidine residue) is as common as the T allele (C = 0.5305) in the AJ population and other populations. However, it is less common in the East Asian population (C = 0.4617) and more common in the African/African American population (C = 0.7001) (gnomAD v4.1.0). Interestingly, previous findings have demonstrated that P2RX7 p.Tyr155 is associated with a gain-of-function in P2RX7 and was indicated to have a protective effect against AD.43,44 This amino acid is located in the extracellular domain of the receptor, 44 within the exposed region of P2RX7 (residues 115–162) that has been implicated in phagocytosis. 45 Phagocytosis is a common pathogenic mechanism in neurodegenerative disorders, 46 including PD, where it contributes to the clearance of α-synuclein and apoptotic neurons. 47 Moreover, defective phagocytosis has been observed in both brain microglia and peripheral blood monocytes of PD patients.47,48 Therefore, we cautiously suggest that P2RX7 p.His155, represented by the alternate allele- C in this study, compromises phagocytic function (compared to the gain-of-function effect of P2RX7 p.Tyr155) and may reduce α-synuclein clearance which might be linked to elevation in PD risk. Additional studies are warranted to elucidate the molecular mechanism of this variant and the role of P2RX7 in phagocytosis.
We showed that P2RX7 Arg276His is associated with PD risk among LRRK2 G2019S carriers. This variant resides within a conserved region and has been shown to result in an increased ATP-induced response, leading to a gain-of-function effect.49,50 In recent years, multiple reviews have examined the involvement of P2RX7 in neurodegeneration and neuroinflammation, establishing that increased activation of P2RX7 leads to neuroinflammatory processes and ROS secretion.33,34,51 In PD, the LRRK2 G2019S mutation induced ROS production and mitochondrial impairment,52,53 which contribute to oxidative stress and subsequent neuronal death. 54 As ROS is a significant mediator in PD, we suggest that individuals carrying both LRRK2 G2019S and P2RX7 Arg276His variants may have higher ROS levels, which may contribute to PD penetrance in LRRK2 G2019S carriers. However, additional studies are needed to determine the functional effect of this variant and this hypothesis.
P2RX7 p.Glu496Ala was associated with earlier AAO among our LRRK2-PD. This variant is located in the carboxyl-terminal tail of the receptor and leads to its LoF. 55 Interestingly, this variant was associated with PD risk in the Chinese Han population. 40 Additionally, studies on human monocytes, collected from individuals homozygous for Glu496Ala, showed that these monocytes failed to induce apoptosis upon ATP exposure, were unable to facilitate mycobacterial killing, and exhibited reduced IL-1β release.56,57 Nevertheless, further functional studies are necessary to understand the association of this gene, and specifically this variant with the LRRK2 gene and pathway.
Our study has some limitations. In the filtering process, to eliminate the possibility of including variants that are not related to PD, we excluded those with only one carrier and thus these variants were not included in the burden analysis. Another limitation is that we restricted our analysis of DEGs to datasets from the SN that were generated using high-throughput sequencing. These stringent filters may have resulted in the exclusion of potential adaptive immune DEGs in PD. Given that PD pathology also manifests in other tissues, future studies should explore DEGs in additional tissues such as the gut and the putamen. Additionally, our analysis was based on datasets generated from postmortem SN tissues, which is currently the only way to study SN in humans. Consequently, our findings likely reflect gene expression changes in the late stages of the disease, and therefore the observed changes in expression may be due to other disease mechanisms rather than direct causative effects of genetic variations within this gene. However, we employed DEGs as an exploratory method to focus on adaptive immune genes associated with PD.
This study focused on a specific ancestry, AJ, therefore, our findings might be limited to this group of individuals. However, it is possible that these variants and possibly others within this gene are also relevant to other populations. Despite this limitation, using this cohort, which is enriched with GBA1 and LRRK2 carriers (about 34%), provided an advantage as it allowed us to conduct analyses in a stratified manner.
In conclusion, our study reinforces the potential involvement of P2RX7 in PD. We further suggested a potential interplay between P2RX7 and LRRK2 and encouraged additional studies to investigate the functional effects of P2RX7 variants.
Supplemental Material
sj-pdf-1-pkn-10.1177_1877718X241296015 - Supplemental material for P2RX7, an adaptive immune response gene, is associated with Parkinson's disease risk and age at onset
Supplemental material, sj-pdf-1-pkn-10.1177_1877718X241296015 for P2RX7, an adaptive immune response gene, is associated with Parkinson's disease risk and age at onset by Shachar Shani, Mali Gana-Weisz, Anat Bar-Shira, Avner Thaler, Tanya Gurevich, Anat Mirelman, Nir Giladi, Roy N. Alcalay, Avi Orr-Urtreger and Orly Goldstein in Journal of Parkinson's Disease
Supplemental Material
sj-tif-2-pkn-10.1177_1877718X241296015 - Supplemental material for P2RX7, an adaptive immune response gene, is associated with Parkinson's disease risk and age at onset
Supplemental material, sj-tif-2-pkn-10.1177_1877718X241296015 for P2RX7, an adaptive immune response gene, is associated with Parkinson's disease risk and age at onset by Shachar Shani, Mali Gana-Weisz, Anat Bar-Shira, Avner Thaler, Tanya Gurevich, Anat Mirelman, Nir Giladi, Roy N. Alcalay, Avi Orr-Urtreger and Orly Goldstein in Journal of Parkinson's Disease
Footnotes
Acknowledgments
Data used in the preparation of this article were obtained on Feb. 24, 2024, from the Accelerating Medicine Partnership® (AMP®) Parkinson's Disease (AMP-PD) Knowledge Platform. For up-to-date information on the study, visit https://www.amp-pd.org. The AMP® PD program is a public-private partnership managed by the Foundation for the National Institutes of Health and funded by the National Institute of Neurological Disorders and Stroke (NINDS) in partnership with the Aligning Science Across Parkinson's (ASAP) initiative; Celgene Corporation, a subsidiary of Bristol-Myers Squibb Company; GlaxoSmithKline plc (GSK); The Michael J. Fox Foundation for Parkinson's Research; AbbVie Inc.; Pfizer Inc.; Sanofi US Services Inc.; and Verily Life Sciences. ACCELERATING MEDICINES PARTNERSHIP and AMP are registered service marks of the U.S. Department of Health and Human Services. Samples were sourced from the Parkinson's Progression Markers Initiative (PPMI), Harvard Biomarkers Study (HBS), Parkinson's Disease Biomarkers Program (PDBP), and the BioFIND study. PPMI is sponsored by The Michael J. Fox Foundation for Parkinson's Research and supported by a consortium of scientific partners: [list the full names of all of the PPMI funding partners found at https://www.ppmi-info.org/about-ppmi/who-we-are/study-sponsors]. The PPMI investigators have not participated in reviewing the data analysis or content of the manuscript. For up-to-date information on the study, visit www.ppmi-info.org. The Harvard Biomarker Study (HBS) is a collaboration of HBS investigators [full list of HBS investigators found at https://www.bwhparkinsoncenter.org/biobank/ and funded through philanthropy and NIH and Non-NIH funding sources. The HBS Investigators have not participated in reviewing the data analysis or content of the manuscript. The Parkinson's Disease Biomarker Program (PDBP) consortium is supported by the National Institute of Neurological Disorders and Stroke (NINDS) at the National Institutes of Health. A full list of PDBP investigators can be found at ![]() . The PDBP investigators have not participated in reviewing the data analysis or content of the manuscript. BioFIND is sponsored by The Michael J. Fox Foundation for Parkinson's Research (MJFF) with support from the National Institute for Neurological Disorders and Stroke (NINDS). The BioFIND Investigators have not participated in reviewing the data analysis or content of the manuscript. For up-to-date information on the study, visit michaeljfox.org/news/biofind. This work was performed in partial fulfillment of the requirements for a PhD degree of Shachar Shani, Faculty of Medicine and Health Sciences, Tel Aviv University, Israel.
. The PDBP investigators have not participated in reviewing the data analysis or content of the manuscript. BioFIND is sponsored by The Michael J. Fox Foundation for Parkinson's Research (MJFF) with support from the National Institute for Neurological Disorders and Stroke (NINDS). The BioFIND Investigators have not participated in reviewing the data analysis or content of the manuscript. For up-to-date information on the study, visit michaeljfox.org/news/biofind. This work was performed in partial fulfillment of the requirements for a PhD degree of Shachar Shani, Faculty of Medicine and Health Sciences, Tel Aviv University, Israel.
Funding
This research was funded by the Chaya Charitable Fund, Michael J. Fox Foundation, and Biogen, Inc.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
The data supporting the findings of this study are available within the article and/or its supplemental material.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.