
Abstract
Low penetrance of Parkinson’s disease (PD) associated with GBA pathogenic variants indicates the presence of modifiers genes. Clusters of PD cases in certain families with GBA variants would serve as a strong evidence for the clinical relevance of such modifiers. We studied eight family trees of non-Parkinsonian, GBA-N370S homozygote, Gaucher probands, with multiple cases of PD. Differences in PD risk associated with different GBA variants were balanced by variant homozygosity. In these families, all PD cases stemmed from only one of the proband’s parents. This observation provides a direct epidemiological evidence for genetic modifiers determining PD risk in GBA variant carriers.
INTRODUCTION
Pathogenic variants in the Glucocerebrosidase (GBA) gene are associated with Parkinson’s disease (PD) [1–3]. The penetrance of these variants is low, and only a fraction of GBA mutation carriers will develop clinically-apparent PD [4]. Reduced penetrance is attributed, at least partially, to genetic modifiers that modulate the GBA gene expression and the enzymatic activity of glucocerebrosidase [5, 6]. However, epidemiological evidence for the relevance of genetic modifiers in reducing or increasing the risk for PD are sparse. To test whether the genetic background of the pathogenic GBA allele modifies risk for PD, we studied how PD cases are distributed in families of obligatory GBA mutation carriers. We hypothesized that families with clusters of PD cases would indicate that genetic elements outside the mutated GBA allele modulate the risk for PD among GBA carriers.
METHODS
We reviewed the family trees of 233 patients with Gaucher disease (caused by biallelic GBA mutations) in the records of the Gaucher Unit, Shaare Zedek Medical Center. This study was approved by the institute IRB committee (0168-16-SZMC) and participants signed informed consent. Families that fulfilled two predefined criteria were included in our analysis: 1) A proband with Gaucher disease (GD), homozygote for the non-neuronopathic GBA-N370S (NM_001005741.2:c.1226A >G) mutation in gene sequencing (Centogene, Rostock), 2) Two or more reported cases of PD among the proband’s blood-related relatives. Affected siblings and offsprings were not included since our aim was to compare the paternal and maternal branches of the family. Individuals younger than 40 years were excluded from analysis since PD onset in this age group is rare.
To calculate the probability that the reported results occurred by chance, we calculated, for each family separately, the probability that the second case of PD in the family would stem from the same branch. This probability was calculated by dividing the number of all individuals in the affected branch minus 1 (to exclude the first case of PD) by the sum of individuals in both the paternal and maternal branches minus 1. In case a third case of PD in the same family existed, the same formula was used and both the first and second cases of PD were excluded from the number of individuals, etc. We then calculated the probability of the reported results by multiplying all these probabilities. Finally, to calculate the P-value for our observed probability, we ran a MATLAB simulation with random PD cases 100,000 times using identical pedigrees and calculated the distribution of random probabilities.
RESULTS
We traced 77 cases of reported PD in 57/233 (24.4%) families. In 15 families, two or more PD cases were reported (up to four cases within a single family). Based on our predefined criteria, we included in our study 19 cases of PD from 8 families of N370S/N370S probands (Fig. 1A-H). The reported median age at PD onset was 63 (range-36–75) years. In these 8 families, all PD cases stemmed from only one of the proband’s parents, although both parents were obligatory N370S mutation carriers. Applying the probability model revealed a p-value for a similar result of 0.0024. These results could also not be explained by an imbalance in the age of individuals in the PD-affected and non-affected sides, median age at the time of death or last follow-up of 71 (range 48–84) years and 66 (range 39–92) years, respectively (Wilcoxon rank sum test p-value = 0.44).
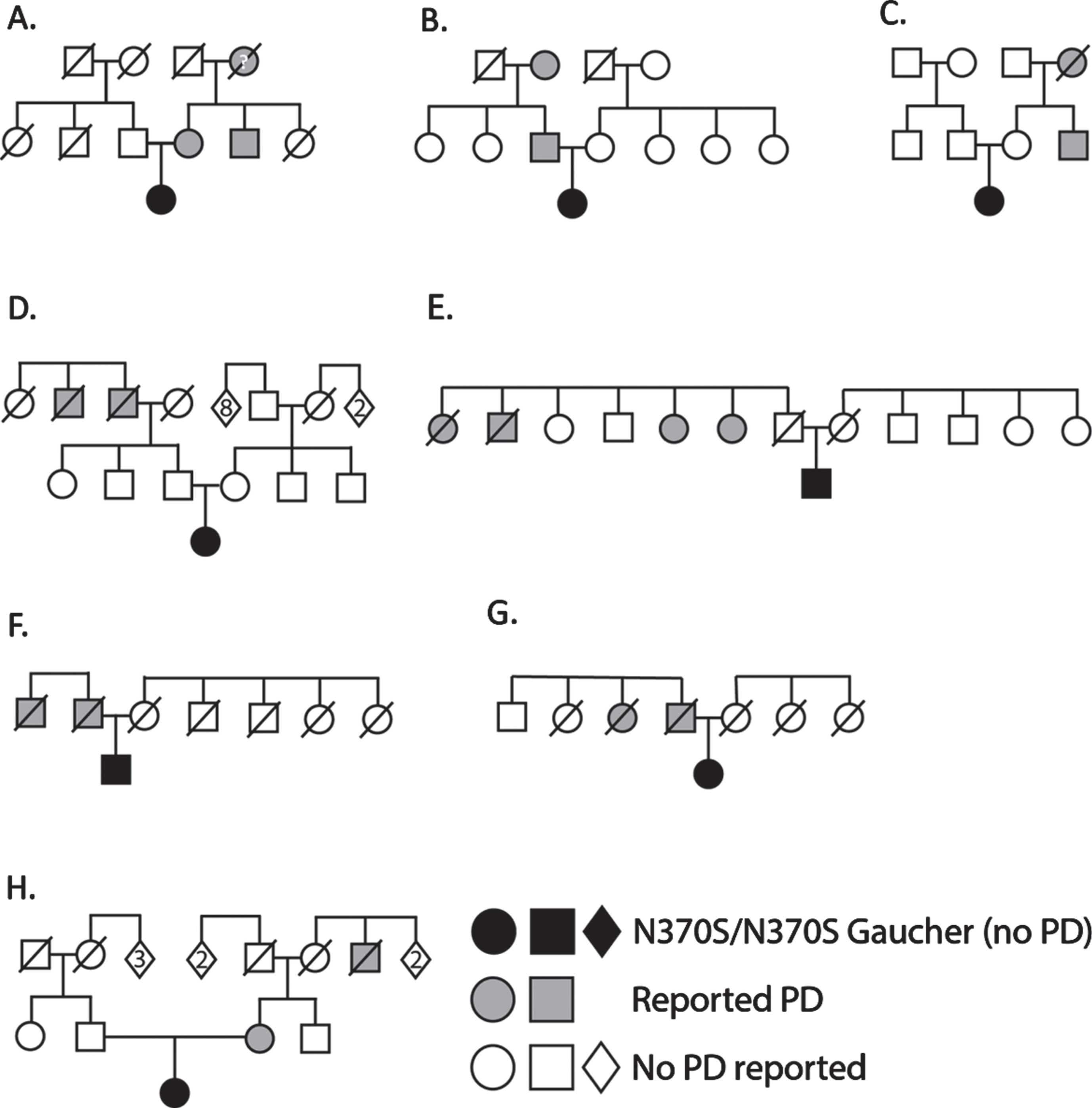
Family trees of GBA N370S homozygous Gaucher probands with multiple Parkinson’s disease (PD) cases. Probands (without PD) are shown in black and PD patients in grey, in eight unrelated families (A-H). In all cases, individuals with PD are clustered in either the maternal or paternal side of the family and never in both. Report of PD in deceased maternal grandmother in family A could not be confirmed due to the absence of documentation.
DISCUSSION
Different GBA mutations are associated with different risks for PD [4, 7]. To homogenize a priori risk probabilities, we focused our research on families of N370S homozygous probands. Carriers of this mutation have a three-fold risk for PD [4]. Where genetic modifiers do not exist, the two branches of such families (paternal and maternal) are expected to carry a similar risk for PD. This was not the case in our cohort. Our results, therefore, strongly support the presence of genetic modifiers that affect the risk for PD in GBA mutation carriers. A literature search for published pedigrees of a homozygous proband with multiple PD cases in the family revealed an additional single family of a proband homozygous to the neuronopathic L444P pathogenic variant [8]. All the members in this family who had PD were from the paternal side. This observation supports our results.
Genetic modifiers that determine the risk for PD among GBA mutation carriers were previously suggested based on findings from in vitro models [5, 6] and a genome-wide association (GWAS) study [5]. The limited clinical relevance of some in vitro studies and the inherited biases of GWAS methodology (see for example [9]) call for more direct evidence for the presence of modifier genes. Our study provides a more direct epidemiological evidence for the existence of such genetic modifiers.
Our study has several limitations. First, cases of PD were reported by the probands and their families but could not be confirmed by physical examination in several cases because some of the individuals were deceased. In such cases, reports were confirmed by consulting medical files. Second, a bias toward detection of PD in families with affected individual, causing the PD cases to look branch-specific, cannot be ruled out. Third, we also did not test the GBA mutation status of the majority of PD cases and, therefore, it is possible that some cases are non GBA-PD. Fourth, we also did not test for the LRRK2 mutation status, as its co-occurrence with GBA mutations is infrequent (around 1%) in the Ashkenazi Jewish population that we examined and, therefore, was unlikely to explain clustering in the vast majority of GBA families. Fifth, the number of PD cases in our cohort is relatively small. This is the result of both the low penetrance of the GBA N370S mutation [4] and the presence of relatively young individuals in our cohort. All these limitations emphasize the need to replicate our observation in additional pedigrees.
Based on our results, we propose that genetic consultants to GBA mutation carriers should take into account the familial occurrence of PD, when assessing their risk of developing PD, in addition to the type of GBA mutation (neuronopathic vs. non-neuronopathic). While a disease-modifying agent for PD has not been identified, clinical trials for such a potential agent should consider carriers with increased risk of PD due to positive familial background as suitable candidates.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.