
Abstract
Chronic sterile inflammation and persistent immune activation is a prominent pathological feature of Parkinson’s disease (PD). Inflammasomes are multi-protein intracellular signaling complexes which orchestrate inflammatory responses in immune cells to a diverse range of pathogens and host-derived signals. Widespread inflammasome activation is evident in PD patients at the sites of dopaminergic degeneration as well as in blood samples and mucosal biopsies. Inflammasome activation in the nigrostriatal system is also a common pathological feature in both neurotoxicant and α-synuclein models of PD where dopaminergic degeneration occurs through distinct mechanisms. The NLRP3 (NLR Family Pyrin Domain Containing 3) inflammasome has been shown to be the primary driver of inflammatory neurotoxicity in PD and other neurodegenerative diseases. Chronic NLRP3 inflammasome activation is triggered by pathogenic misfolded α-synuclein aggregates which accumulate and spread over the disease course in PD. Converging lines of evidence suggest that blocking inflammasome activation could be a promising therapeutic strategy for disease modification, with both NLRP3 knockout mice and CNS-permeable pharmacological inhibitors providing robust neuroprotection in multiple PD models. This review summarizes the current evidence and knowledge gaps around inflammasome activation in PD, the pathological mechanisms by which persistent inflammasome activation can drive dopaminergic degeneration and the therapeutic opportunities for disease modification using NLRP3 inhibitors.
INTRODUCTION
Chronic immune and inflammasome activation triggered by accumulating misfolded protein aggregates is emerging as a key pathogenic mechanism that drives neurodegeneration in Alzheimer’s disease (AD), Parkinson’s disease (PD), and other progressive neurological disorders that are accompanied by chronic unresolving immune activation [1, 2]. Inflammasomes are multi-protein complexes that primarily function as intracellular sensors of environmental, metabolic, or cellular stress. There are multiple inflammasomes such as NLRP1, NLRP2, NLRP3, NLRC2, and AIM2 which are known to be expressed in the CNS in immune, glial, and neuronal cell types. Each of these NLR-family proteins appear to sense diverse biological stimuli ranging from misfolded protein aggregates to bacterial components during CNS infection or viral DNA. The NLR Family Pyrin Domain Containing 3 (NLRP3) inflammasome is the most highly characterized member in PD and consists of the sensor NLRP3, the signaling adapter ASC, and the caspase-1 protease [3]. NLRP3 inflammasome activation is a two-step process: priming and activation. Priming occurs as a result of pattern recognition receptor signaling activated by various pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), activating the NFκB pathway [4]. Activation can be caused by various stressors, including ATP, misfolded proteins, or particulate matter, leading to NLRP3 complex assembly; activation is generally accompanied by potassium efflux, mitochondrial dysfunction, and cathepsin B release from lysosomes [5, 6]. Assembly of the NLRP3 complex in peripheral and brain immune cells drives autoproteolytic activation of caspase-1 and subsequent release of interleukin-1β (IL1β), interleukin-18 (IL18), and Gasdermin D (GSDMD). The N-terminal fragment of GSDMD forms a pore in the plasma membrane, allowing proinflammatory cytokines and other intracellular materials to leak out [3]. The acute and transient activation of the inflammasome is typically a protective response necessary for overcoming infection or other physiological stressors. Chronic unresolving inflammasome activation, however, appears to play a pathological role in the progression of neurodegenerative disorders [7, 8]. In PD, persistent NLRP3 inflammasome activation in innate immune cells is triggered by accumulating misfolded α-synuclein and leads to dopaminergic (DA) neuronal cell death through the release of proinflammatory cytokines. There is a direct link between chronic inflammasome activation in the CNS and inflammasomes driving the pathology and further propagation of protein aggregates [9].
Studies from independent groups have now confirmed that markers of inflammasome activation are strongly evident in the central nervous system (CNS) at the sites of neurodegeneration, as well as in the blood of patients with AD, PD, and other neurological disorders such as amyotrophic lateral sclerosis [10]. Ground-breaking studies in AD have demonstrated that extracellular inflammasome components such as prionoid ASC can indeed cross-seed and propagate misfolded pathological proteins and thereby drive neuropathology [11]. From a therapeutic perspective, inflammasome activation is now considered to be one of the most promising targets for disease modification in the CNS. This is based on evidence in APP/PS1 AD models showing that NLRP3 knockout mice can clear pathogenic amyloid plaques, and pharmacological studies in MitoPark, 6-hydroxydopamine, and α-synuclein preformed fibril (PFF) PD models demonstrating that NLRP3 inhibition in the CNS can prevent α-synuclein pathology and dopaminergic degeneration [2, 13]. Further, the single nucleotide polymorphism (SNP) in NLRP3 rs7525979 correlates with a statistically reduced risk of PD as the polymorphism impacts NLRP3 translation, resulting in a protein product with decreased stability and solubility compared to WT NLRP3 [14]. This review summarizes current knowledge around inflammasome activation during PD and its pathophysiological roles in disease progression highlighting current knowledge gaps and therapeutic strategies for disease modification through inflammasome inhibition.
SYSTEMIC AND CNS INFLAMMASOME ACTIVATION IN PD
A defining pathological hallmark of progressive neurodegenerative diseases is an accumulation and spread of insoluble proteins unique to each condition, such as Lewy body aggregates in PD. Chronic activation of the innate immune system in response to these persistent protein aggregates is emerging as a central pathological mechanism that can drive progressive neurodegeneration. Since the early reports of elevated inflammatory markers in PD patients were made by McGeer and others [15], a wealth of evidence from clinical studies, pre-clinical models and mechanistic data supports the paradigm that sustained neuroinflammation can exacerbate ongoing neurodegeneration and drive disease progression.
Clinical studies and epidemiological data on inflammasome activation markers in PD
Extensive inflammasome activation has been detected in the postmortem brains of PD patients, with elevated NLRP3 expression at both protein and mRNA levels [12, 16]. In a cohort of 17 PD patients, histological studies indicated significantly higher expression of NLRP3 in the substantia nigra compared to 11 healthy controls; over 50% of cells were NLRP3 positive compared to only 20% of cells in healthy controls. Complementary mRNA data show an approximate 2-fold increase in gene expression of NLRP3 in mesencephalic tissues of PD patients [14]. Similar results are observed in histological studies of ASC expression, with over twice as many cells expressing ASC in postmortem mesencephalic tissues of PD patients [16]. Additionally, significant increase, 1-fold, in protein expression of cleaved caspase-1 p20 and ASC is observed in PD substantia nigra tissue compared to aged matched healthy controls [12].
In addition to the presence of NLRP3 and inflammasome markers in brain tissues, there is also evidence of widespread expression of inflammasome markers in bloods of PD patients. Serum samples of 27 PD patients had significantly elevated IL1β (45.53±15.06 pg/mL versus 219.83±58.59 pg/mL) and NLRP3 (6.97±1.44 ng/mL versus 33.56±5.05 ng/mL) compared to 15 healthy controls. These elevated NLRP3 protein levels also correlate linearly with increased expression of α-synuclein (Spearman’s rho 0.843) [17]. Similarly, plasma samples of PD patients have significantly increased protein levels of oligomerized α-synuclein (0.171±0.044μM/L versus 0.676±0.083μM/L), phosphorylated α-synuclein (0.128±0.041μM/L versus 0.849±0.108μM/L) and IL1β (0.604±0.136μM/L versus 1.876±0.327μM/L) compared to their healthy counterparts [18]. Another study indicated significantly elevated protein expression of pro-IL1β, p20, pro-GSDMD, and cleaved GSDMD is present in PD patient extracellular vesicles (EVs), suggestive of pyroptosis-associated vesicular shedding. EV-borne NLRP3 was detectable in 33% of the 31 PD patient samples tested compared to 18% of the 11 healthy control samples tested [16].
Inflammasome activation in preclinical animal models of PD
Elevated inflammasome activation markers have been confirmed in most of the commonly used models of PD to date (Table 1), being strongly evident in the nigrostriatal system, blood, and gastrointestinal tract, similar to findings in PD patients. Importantly, inflammasome activation appears to be a common underlying theme in all pre-clinical PD models irrespective of the different pathological triggers used to initiate these models ranging from α-synuclein aggregates, dopaminergic neurotoxicants such as 1-methyl-4-phenyl-1,2,3,6-tetrahrdyopyridine (MPTP) or PD-linked environmental chemicals such as paraquat (PQ) or manganese (Mn) [12, 19]. These findings have emerged from independent studies across multiple research groups, providing credence to the inflammasome activation paradigm in multiple PD models. This highlights that multiple triggers of neuroinflammation and neuropathology in PD converge on inflammasome activation as a common pathological mechanism.
Inflammasome activation in clinical and experimental Parkinson’s disease
Two commonly used neurotoxicants for modelling PD are 6-hydroxydopamine and MPTP. Both 6-OHDA and MPTP have affinity for the dopamine transporter, which allows for a level of specificity for dopaminergic neurons [20]. Treatment with 6-OHDA induces reactive oxygen species (ROS) which in turn activates NLRP3 expression and results in the production of inflammasome markers IL1β and IL18 [21, 22]. In the 6-OHDA model, strongly elevated inflammasome activation markers including p20, ASC and NLRP3 are evident early in the disease course at 3 days, suggesting that NLRP3 activation precedes loss of dopaminergic neurons. Further, the upregulation of NLRP3 and ASC were found to be almost exclusively in hypertrophic reactive microglia-like cells which were Iba1-positive [12]. In the MPTP model, pro-IL1β mRNA expression in the substantia nigra of MPTP treated mice is upregulated 5-fold [23]; NLRP3 protein expression is upregulated and observed to localize to microglia and astrocyte cells [24]. Treatment with lipopolysaccharide (LPS)/MPTP in three day mouse studies results in significant upregulation of protein expression of ASC specks, IL1β, and NLRP3 in SN tissue, while treatment with NLRP3 inhibitors returns the aforementioned protein levels to closer to control values [25].
Pesticides PQ, rotenone, and Mn have all been used to model inflammasome activation in studying PD. Pesticide exposures have been linked to oxidative stress and mitochondrial dysfunction which are upstream triggers of inflammasome activation and the production of inflammasome mediators in immune cells. PQ produces a large amount of ROS as it undergoes a redox cycling process, leading to NFκB and inflammasome activation [26]. Additional treatment with maneb, a fungicide, leads to dopaminergic neuronal death and exaggerates oxidative stress induced by treatment with PQ [27]. 8-week studies of PQ and maneb-induced mouse models of PD resulted in 1 and 2-fold higher protein expression of α-synuclein and NLRP3, respectively, while parallel treatment with inflammasome inhibitors reduced NLRP3 expression back to that of the control [28]. Rotenone causes pyroptotic cell death through activation of caspase-1 and NLRP3. 24-hour treatments of 4μM rotenone on HT22 cells results in significantly elevated ROS levels and increased caspase-1, NLRP3, and IL18 protein expression [29]. 6-month rotenone-induced PD mouse model, expression of tyrosine hydroxylase (TH)-positive neurons was significantly reduced following treatment; NLRP3 knockouts, however, did not see a comparable loss, highlighting the role of NLRP3 in rotenone activity [30].
Mn is a neurotoxic metal, and excessive chronic exposure results in manganism. Manganism is a condition which presents with parkinsonian features, including generalized bradykinesia and rigidity [31]. Unlike PD, manganism is seldom responsive to treatment with levodopa, and presents with a resting tremor less often [32, 33]. Mn exposure is linked to protein misfolding, mitochondrial impairment and neuroinflammation [34]. Sarkar et al. determined that Mn-primed microglial cells increased expression of NLRP3, and that this effect was transferrable to other cells via purified ASC-containing exosomes released from Mn-primed microglia. This finding was corroborated in Mn-exposed welders; serum exosomes in this cohort contained significantly higher concentrations of ASC compared to healthy controls [35]. In BV2 cells, ROS is higher in Mn-treated cells as determined by flow cytometry. A dose dependent increase in protein expression of NLRP3 is observed in cells treated with 100 to 400μM/L MnCl2 for 24 h. In Mn-treated rats, increase in protein expression of IL1β, IL18, and NLRP3 in the basal ganglia were observed after 8 weeks of dosing 5 days a week [36].
In addition to the Parkinsonian neurotoxicant and PD-linked pesticide models, inflammasome activation has been shown to occur in multiple α-synuclein models of PD which model hallmark Lewy body pathology that occurs in PD. α-synuclein PFF has been shown to initiate inflammasome activation, with microglial NLRP3 detected via immunostaining after 30 days. Further, ASC and p20 were detectable in the striatum of α-synuclein PFF mice [12]. α-synuclein transgenic mouse lines over express α-synuclein and are widely used to model systemic and non-motor PD pathology. At 2 months and 12 months, α-synuclein transgenic mice show increased pro-IL1β and IL1β mRNA expression in substantia nigra tissue [23]. A53T transgenic mice have increased NLRP3, caspase-1, pro-IL1β, and IL1β protein expression in midbrain tissue, regardless of MPTP treatment [37].
PRIMING AND ACTIVATION OF THE NLRP3 INFLAMMASOME IN PD
While widespread inflammasome activation has been confirmed in both human PD and preclinical animal models (Table 1), the mechanisms by which priming and activation of the inflammasome are triggered and/or become dysregulated remain poorly defined. In most immune cells, activation of NLRP3 is closely regulated and requires an essential ‘priming’ step, which facilitates NFκB-mediated upregulation of the various inflammasome components that are necessary to license assembly of the inflammasome complex when the relevant activation signal is present [48]. Although the mechanisms by which the inflammasome priming and activation occurs in PD have not been fully elucidated, emerging evidence suggests that many of the known pathological triggers and mechanisms linked to PD are likely to be key players in this process.
Binding of α-synuclein aggregates to Toll-like receptors (TLRs) has been shown to be an important trigger for microglial activation [49], which could also be an important step in priming of the NLRP3 inflammasome. NLRP3 priming via translocation of NFκB is not unique to TLRs; multiple signaling cascades converge at this point, including other pattern recognition receptors, tumor necrosis factor receptor (TNF-R), and interleukin-1 receptor (IL-1R) [50, 51]. In organotypic hippocampal slices, activation of TLR2 and TLR4 primes the inflammasome, causing increased expression of NLRP3 and IL1β [52]. ASC specks were detectable with the addition of nigericin, imiquimod, or ATP as the activation signal in these studies [52]. In the MPTP model, TLR4 knockout mice exhibited decreased loss of TH-positive cells, while protein expression of IL1β, caspase-1, and NLRP3 were decreased in the knockout compared to WT, indicating that TLR4 could be involved in priming of the NLRP3 inflammasome for activation in PD [53].
NLRP3 knockout mice treated continuously with MPTP for 28 days did not display nigral dopaminergic degeneration or striatal dopamine deletion compared to their WT counterparts. Additionally, there was prevention of α-synuclein aggregate formation in NLRP3 knockouts compared to WT [43]. NLRP3 deficient mice exhibit significantly decreased motor symptoms and dopaminergic neuronal degeneration in the MPTP model of PD. Additionally, microglia activation, IL1β production, and caspase-1 activation were all reduced in knockout mice. MPTP and ATP treatments of glial cultures cause the production of mitochondrial ROS, ultimately leading to the assembly and activation of NLRP3, where MPTP appears to play a role in inflammasome priming [42]. Primary astrocytes treated with varying doses of α-synuclein resulted in increased expression of NLRP3, caspase-1, and IL1β in a dose-dependent manner. Treatment with the autophagy inhibitor 3-MA suppressed NLRP3, caspase-1, and IL1β expression [18]. Deletion of autophagy regulated gene 5 (Atg5) in a mouse model resulted in motor coordination impairment, loss of TH neurons, neuroinflammation, and decreased dopamine levels in the striatum. Mechanistic studies showed activation of NLRP3 resulting from the Atg5 knockout which led to increased expression of macrophage migration inhibitory factor (MIF); MIF is upregulated in PD patients [54]. miR-29c-3p is protective against inflammation in MPTP-treated animal and LPS-induced PD microglia cell culture models; it was observed to be down regulated in BV-2 cells primed with LPS, while NFAT5 knockdown showed similar results. Conversely, the overexpression of miR-29c-3p conferred protection with decreased levels of IL1β, NFkB, and NLRP3 activation [55]. Parkin, a PD-linked protein associated with protein metabolism and mitochondrial function has also been linked to priming and activation of the inflammasome in PD. Knockdown of Parkin in both sporadic PD mouse model and human embryonic stem cell differentiated DA neurons show priming and activation of NLRP3. Overexpression of Parkin in caspase-1 knockout mice did not observe the same results, as adulthood Parkin knockdown did not cause DA neuronal cell death. Parkin was inactivated in the α-synuclein PFF model, suggesting disinhibition of NLRP3 inflammasome licensing leads to DA neuronal death [56].
Immune cell subsets linked to inflammasome activation in PD
Microglia are the primary immune cell in the CNS and play a critical role in maintaining a healthy microenvironment within the brain. In the diseased state, however, microglia drive neuroinflammation and inflammasome activation [57]. α-synuclein aggregates play a key role in microglial activation in PD. TLR2 and TLR5 ligation with α-synuclein causes microglia to engulf the aggregates and leads to NLRP3 activation [58]. The regulation of the uptake of α-synuclein in human microglia has been shown to be regulated by Fyn kinase and class B scavenger receptor CD36. Fyn kinase also regulates microglial NLRP3 priming and activation; priming is controlled by the translocation of NFκB while the phagocytosis and lysosomal rupture triggered by α-synuclein fibrils activates ROS leading to inflammasome activation [59]. Interestingly, microglia do not degrade α-synuclein oligomers while the inflammasome is activated; inhibition of NLRP3 however, facilitated clearance of α-synuclein oligomers by microglia [58]. LPS primed microglia display an increase in NLRP3 as well as pro-IL1β production. Inhibition of mitochondrial complex-1 increased ASC speck formation, turnover of pro-IL1β to IL1β and NLRP3 activation, highlighting the role of mitochondrial impairment in inflammasome activation. Additionally, primary mesencephalic and human dopaminergic neuronal cells underwent dopaminergic neurodegeneration when exposed to the supernatants of activated microglia cells [45]. In contrast, NLRP3-/- murine microglia did not produce IL1β in response to α-synuclein as opposed to their wildtype counterparts [60]. This highlights the upstream requirements for inflammasome assembly prior to the production of IL1β in microglia activated during PD. NLRP3-induced secretion of IL1β from cultured primary microglia has been validated in multiple human and murine models of PD [12, 61].
In a cohort of 43 PD patients and 24 healthy controls, NLRP3 expression was elevated in activated peripheral blood mononuclear cells (PBMCs) of the PD patients, indicating increased caspase-1 and mature IL1β compared to internal controls such as GAPDH. This indicates that peripheral immune cells are primed in PD for activation of the NLRP3 inflammasome. Additionally, α-synuclein levels in PD patients’ blood are higher compared to healthy controls; when compared, there was a positive correlation between α-synuclein in the blood and the increase of inflammasome markers in PD patient PBMCs. This was additionally correlated to the Hoehn and Yahr staging scale which reflects on PD progressive symptoms [39].
Platelets play a key role in inflammasome regulation, boosting NLRP3 activation in macrophage cells. In cell cultures, co-culturing of human macrophages, neutrophils, or monocytes with platelets significantly increased IL1β production resulting from a combination of LPS, ATP, and Nigericin treatments. Though not yet fully understood, NLRP3 transcription and inflammasome activation is enhanced because of factors released by platelets [62].
TRIGGERS AND DRIVERS OF PERSISTENT INFLAMMASOME ACTIVATION IN PD
There are numerous triggers that lead to the assembly and activation of the inflammasome (Fig. 1). A key pathway is the activation of NFκB which primes the inflammasome for upregulation of proinflammatory cytokines. This step enables inflammasome assembly when the second activation stimulus is introduced. This section will focus on a discussion of the primary triggers associated with inflammasome activation in PD.
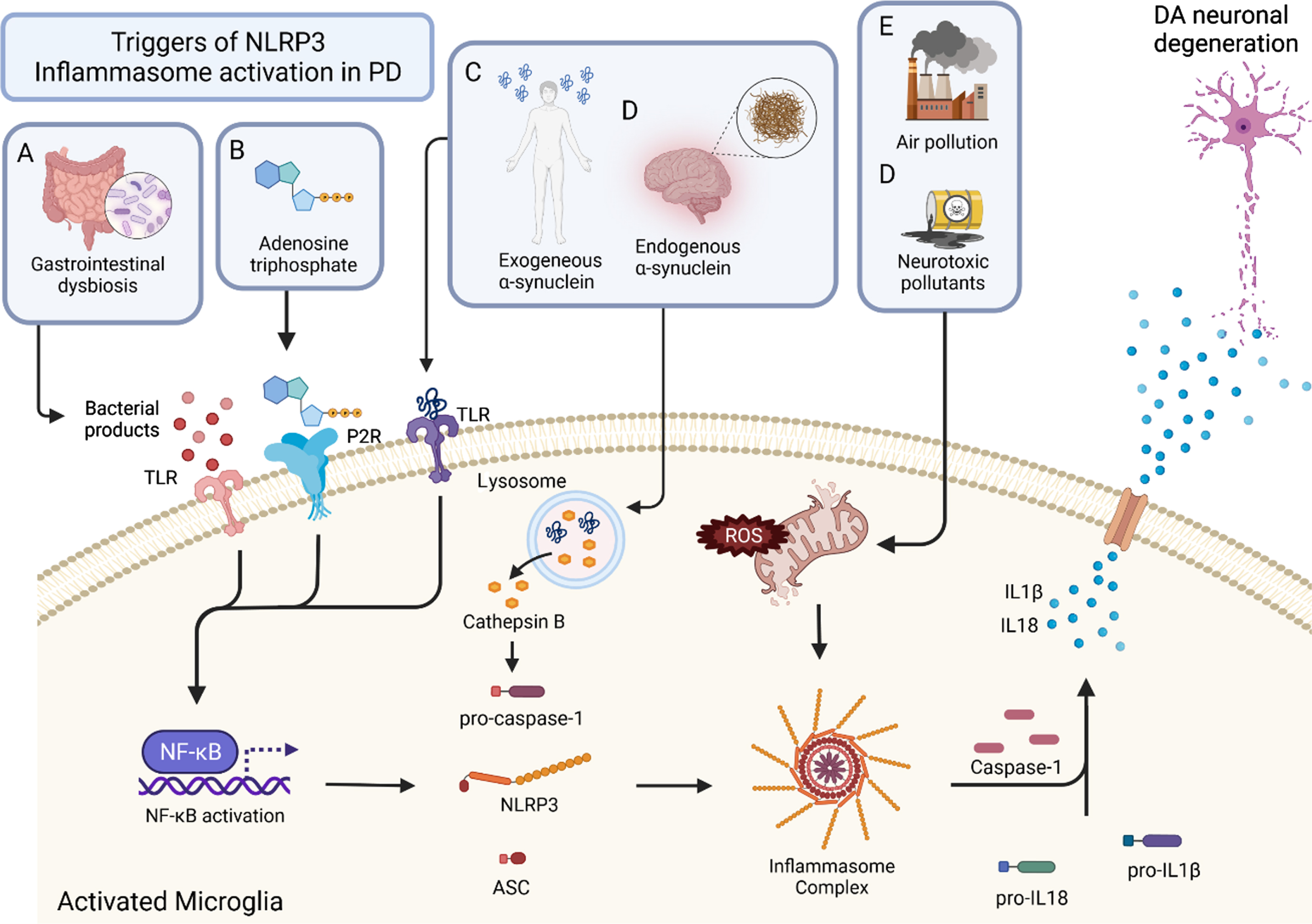
Triggers and mechanisms of inflammasome activation in PD. Activation of the inflammasome can come from a variety of sources. Triggers of the inflammasome A, B, and C result in the activation of NFκB. This priming step leads to the production of NLRP3, ASC, pro-caspase-1, pro-IL18, and pro IL1B. With activation, the inflammasome complex is formed, which cleaves pro-caspase-1 to the active form, caspase-1. Caspase-1 then cleaves the pro-forms of IL1β and IL18 to give their mature, active forms. These inflammatory cytokines are secreted from the microglia and lead to DA neuronal degeneration. Bacterial products of gastrointestinal dysbiosis (A) interact with TLRs which activate NFκB. Adenosine triphosphate (ATP) (B) activates P2Rs leading to NFκB activation. Exogenous α-synuclein (C) and endogenous α-synuclein (D) interact with TLRs as well, leading to the activation of NFκB. Air pollution (E) and neurotoxic pollutants (F) cause mitochondrial stress and result in the production of ROS which leads to inflammasome formation and ultimately inflammatory cytokine production.
Pathological α-synuclein aggregates
α-synuclein is critical to the development of PD; it is the primary component of Lewy body aggregates detectable in postmortem brain sections of PD patients [63]. Misfolded α-synuclein functions as a prion, with exosomes facilitating transport for synuclein aggregates between neuronal cells. Moreover, interfering with exosome secretion from microglia cells prevented the subsequent migration of α-synuclein to neurons [64]. It has been speculated that NLRP3 activation facilitates the transport of α-synuclein via exosomes [65]. Mn stimulation causes cells expressing wildtype human α-synuclein to secrete exosomes containing misfolded synuclein leading to neuroinflammation after uptake of exosomes by microglia [66]. α-synuclein has been shown to induce NLRP3 activation in multiple murine and human microglia models leading to the production of IL1β [12, 60]. The mechanisms at play in each instance, however, vary slightly as it does not appear that caspase-1 activity is critical for IL1β secretion in human microglia where it is in murine microglia. Treatment with caspase-1 inhibitor Z-YVAD-FMK prevents IL1β production associated with α-synuclein mediated inflammasome activation in mouse microglia; the same treatment in human microglia did not block IL1β production suggesting caspase-1 may not be necessary for IL1β maturation in human microglia [60].
In human induced pluripotent stem cell derived microglia (hiMG), α-synuclein activated NLRP3, while treatment with α-synuclein antibody resulted in increased secretion of IL1β. In mouse brain grafts of α-synuclein, caspase-1 is activated, and neurotoxicity is observed. Again, α-synuclein antibodies exacerbated these reactions, suggesting that the protein in any form can agitate and initiate inflammasome activation [67]. Several studies from independent groups have now established that misfolded synuclein fibrils are a key trigger for NLRP3 activation in peripheral and CNS-resident immune cells. Crucially, pharmacological inhibition of NLRP3 or genetic ablation in immune cells, almost completely abolishes the production of inflammasome mediators, suggesting that other NLR proteins and pathways likely do not contribute to inflammasome activation in these cells.
ATP
ATP is a common stimulant used to study inflammasome activation in cell models as it is released from dying or injured neurons. ATP exposures have been directly linked to NLRP3 inflammasome activation in cultured macrophages. Glutathione (GSH) efflux is stimulated after ATP exposure, as GSH levels were elevated in supernatants. Zhang et al. propose a link between glutathione and NLRP3 activation, where glutathione levels control inflammasome activation [68]. P2X4 receptors, triggered by extracellular ATP, were activated in rats injected with 6-OHDA. IL1β and IL18 were also elevated, suggesting NLRP3 activation could be involved. When treated with a P2X4-specific receptor agonist, activation of NLRP3 was alleviated. The same result was obtained when tested in P2X4 knockouts. P2X4R antagonism as well as siRNA-mediated P2X4R knockdown alleviated NLRP3 inflammasome activation in a 6-ODHA rat model of PD. In addition, purinergic receptors have also been implicated in DA homeostasis; in a 6-OHDA model of PD, P2X4R-knockout mice displayed significantly decreased levodopa-induced behavioral response which may have been caused by altered nigrostriatal signaling [22].
Neurotoxicants and chemical exposures
Exposure to pesticides and other chemical exposure is a known cause of PD. Individuals in certain high-risk occupations, such as farmers, have up to a 16% higher rate of PD occurrence compared to the general population [69]. The use of various pesticides and their link to PD and its progression have been previously reviewed [19, 71]. Treatment with Mn results in impaired mitochondria function in microglia cells, leading to inflammasome activation [35]. Additionally, Mn exposure increases inflammation in peripheral blood and in the CNS. Healthy rat gut microbiota transplanted into the guts of rats who had Mn exposure resulted in a downregulation of NLRP3 expression as well as other neuroinflammatory factors, highlighting the important role of the gut microbiome in NLRP3 inflammasome regulation [46].
Electron transport chain defects and mitochondrial dysfunction occur early in the natural history of PD [72]. Postmortem analysis of PD patient brain tissue indicated loss of complex-1 function, an integral component of the respiratory chain [73]. In mice with induced complex-1 dysfunction, a progressive loss of dopaminergic neurons was observed, beginning in nigrostriatal axons [74]. Rotenone and tebufenpyrad are mitochondrial complex-1 inhibitors, and LPS-primed microglial cells specifically amplified NLRP3 response upon exposure to these pesticides. Rotenone and tebufenpyrad enhanced ROS generation in primary microglia, whereas treatment with mito-apocynin, a mitochondria-targeted antioxidant, completely inhibited IL1β release [45]. As with 6-OHDA treatment discussed earlier, this pathway converges upon ROS-induced activation of NLRP3. These pesticides are known to result in the production of bioenergetic defects and lysosomal dysfunction. Animal models that undergo chronic rotenone exposure also suggest impairment of mitochondrial function associated with NLRP3 activation [45]. Murine hippocampal HT22 cells incubated with rotenone for 24 h had significantly increased protein expression of NLRP3, caspase-1, IL18, and IL1β [29]. PQ acts by multiple mechanisms that are not yet fully understood. However, it has been shown to activate the NLRP3 inflammasome leading to the production of inflammasome-associated cytokines [8, 75–77]. PQ exposure has also been linked to a higher risk of PD onset in humans and this has been demonstrated in PQ-poisoning rat models [78].
An intriguing link between air pollutants and increased risk of developing PD has been postulated based on clinical, epidemiological, and mechanistic animal studies [79–85]. Particulate matter, carbon monoxide, and metals, all classified as different pollutants, have been shown to be elevated in the brain of individuals in urban compared to rural locations, whilst also having mechanistic links to inflammasome activation [86]. NLRP3 is known to be a sensor of particulates with monosodium urate being a well-known particulate activator. Emerging evidence implicates inflammasome priming, assembly, and activation by particulate air pollutants. Indeed, exposure to fine particulates found in air pollution (≤2.5μm) which increase the risk of PD in some studies have also been shown to increase NLRP3 inflammasome activation markers leading to atherosclerosis [85, 87]. Likewise, diesel exhaust particles which are linked to an increased risk of PD, are also known to be triggers of inflammasome activation in other diseases. However, these links have not been demonstrated in PD models or patient studies [88].
Gut dysbiosis
As the mucosa of the gastrointestinal tract is continuously exposed to foreign antigens, low levels of inflammation are tonically present even in healthy individuals [89]. Thus, a delicate balance between pro- and anti-inflammatory influences is required to maintain mucosal homeostasis and intestinal barrier function. In PD patients, numerous factors tilt the inflammatory equilibrium towards a pro-inflammatory state via activation of the inflammasome. Taken together, clinical and experimental evidence currently suggests the existence of a microbiome-inflammasome axis in PD pathophysiology, acting via TLRs and microbiome metabolite receptors to enable modulation of inflammasome assembly. Inflammasome assembly itself appears to enable further Parkinsonian progression, and given the prodromal nature of PD-associated gut dysfunction, may lend credence to Braak’s hypothesis of vagal gut-brain transmission of α-synuclein [90, 91]. Clinical evidence in PD patients indicates a loss of short chain fatty acid (SCFA)-producing genera, alongside proliferation of archetypally pathologic genera [92, 93]. SCFAs are produced by bacterial fermentation of indigestible dietary fiber and are known to regulate activation of NFκB via inhibition of histone deacetylases (HDACs) or via G-protein-coupled receptors (GPCRs) such as GPR109A, GPR41, and GPR43 [94, 95]. Cellular models of the intestinal epithelium, murine models of dextran sulfate sodium (DSS)-induced colitis, and human studies have all shown SCFAs preserve intestinal barrier function via inhibition of the NLRP3 inflammasome [93, 97]. Increased intestinal permeability has been observed in PD patients when compared with healthy controls. PD-related intestinal hyperpermeability was further associated with a commensurate increase in mucosal α-synuclein content and increased serum LPS-binding protein (LBP), a marker for exposure to LPS [98]. In turn, LPS-induced loss of intestinal barrier function has been shown to increase permeability to α-synuclein which appears to cause pathological amyloidogenesis of α-synuclein within the intramural plexuses of the gut [99]. LPS further interacts with the host inflammasome by binding to TLR4, which is overexpressed in the colonic lamina propria of PD patients. TLR-knockout mice were found to resist rotenone-induced intestinal barrier damage; rotenone-induced neuroinflammation and neurodegeneration observed in wild-type mice were also partially ameliorated [40].
Aging
Aging has long been identified as the single greatest risk factor for the development of PD [100]. With age, individuals exhibit increased levels of inflammatory cytokines, a process termed inflammaging, suggesting an increase in basal inflammatory levels with age [101]. NLRP3 activation is a tightly regulated process; however, as we age, these regulations slowly become disorganized and NLRP3 activation becomes more pronounced. Features like autophagy become compromised as autophagic marker counts decrease in PBMCs [102]. Further, aging is associated with enhanced microglial dysfunction; the microenvironment of the brain changes and microglia take on a proinflammatory role, producing inflammatory mediators such as CD68, IL1β, and MHC-II [103, 104].
HOW CHRONIC INFLAMMASOME ACTIVATION DRIVES NEUROPATHOLOGY IN PD
The presence of elevated systemic and central inflammasome activation markers have now been confirmed in PD patients and disease models in multiple clinical, preclinical, and mechanistic studies from independent groups. However, the mechanisms by which inflammasome activation can drive neuropathology are less clear and this understanding could enable the development of more effective therapeutic strategies for disease modification. While the earliest triggers of inflammasome activation in PD are still unknown and difficult to conclusively determine, current evidence suggests that misfolded synuclein aggregates, mitochondrial dysfunction and impaired phagocytic clearance via the lysosome could be some of the mechanisms by which inflammasome activation is triggered in PD.
Current evidence suggests that key pathological mechanisms by which inflammasome mediators drive neuropathology and disease progression range from exacerbation and cross-seeding of protein misfolding and aggregation of synuclein pathology to direct neurotoxicity. As discussed earlier, the impaired ability of microglia to clear synuclein aggregates could be play an important role in PD pathology similar to other neurodegenerative diseases that are characterized by accumulation and spread of these proteins [58]. Additionally, α-synuclein accumulation drives neuropathology, playing a role in both priming and activation of NLRP3 via TLR-2 and TLR-4. Prionoid ASC specks allow for cross-seeding of amyloid-β in the APP/PS1 AD model, with similar mechanisms likely at play with Lewy body pathology in PD [11].
Inflammatory mediators including ASC, caspase-1, IL1β, IL18, and IL-33, trigger and amplify inflammasome neuropathology in PD. In Mn-treated microglia, the transfer of ASC via exosomes has been observed; primed microglia cells treated with exosomes from animals that had been treated with Mn resulted in elevated IL1β expression, suggesting an inflammasome-triggering role for ASC [35]. Caspase-1 is another critical component of the NLRP3 inflammasome, involved in microglia activation leading to PD pathology; caspase-1 knockouts treated with MPTP/probenecid showed decreased DA neuronal loss [105]. Caspase-1 has been shown to cleave α-synuclein to a highly aggregation-prone form, further exaggerating disease pathology. Increased caspase-1 concentrations, varying from 0.5 to 2.5μM, proportionally increases the amount of truncated α-synuclein; MALDI-TOF mass spectrometry analysis was used to determine that cleavage was occurring at ASP121. Aggregation studies indicated aggregation of the truncated form of α-synuclein after just 5 h, compared to approximately 30 h for the full-length α-synuclein protein [106]. The interleukin-1 (IL-1) family is composed of 11 different members, all of which serve unique biological functions [107]. Notably, IL1β, IL18, and IL33 could play key contributory neurotoxic roles in PD pathology. Once the pro-form of these mediators has been proteolytically activated, they contribute to an unresolving inflammatory microenvironment which can lead to progressive neurodegeneration.
There are mixed results around the role of dopamine relative to inflammasome activation. Yan et al. showed that dopamine treatments prior to the addition of nigericin prevented NLRP3-dependent caspase-1 activation, as well as IL18 and IL1β production, suggesting NLRP3 inhibition [108]. Liu saw similar results, showing dopamine at doses 10-3, 10–4, and 10–5 inhibits the production of inflammatory mediators such as IL1β in LPS primed RAW264.7 cells [109]. However, metabolism of dopamine generates products such as hydrogen peroxide and quinones. Inhibition of vesicular monoamine transporter led to increased intracellular concentrations of dopamine and ultimately dopamine autooxidation [20]. Activation of NFκB via dopamine leads to priming of NLRP3 in primary human macrophages [110].
Mitochondria dysfunction is a recognized pathological hallmark of PD which also has mechanistic links to inflammasome activation. Pathways regulating cellular bioenergetics that are expressed in response to peroxisome proliferator–activated receptor γ coactivator-1α (PGC-1α) have been shown to be lower in people with PD [111]. While these findings were reported over a decade ago from large genome-wide expression studies in dopaminergic neurons from over 400 PD patient brain samples, more recent studies have confirmed that PGC-1α is also a negative regulator of inflammasome activation, suggesting that this pathway functions as an endogenous brake which keeps inflammasome activation in check [45, 112]. Similarly, increased oxidative damage and ROS have been observed in PD patients and multiple animal models while both mitochondrial and cellular ROS can prime or activate the NLRP3 inflammasome [113]. The intriguing overlap in pathological mechanisms linked to PD and NLRP3 inflammasome activation is also evident in the fact that the major environmental toxicants linked to a higher risk of PD have also been shown to be triggers of NLRP3 activation.
THERAPEUTIC TARGETING OF INFLAMMASOMES FOR DISEASE MODIFICATION IN PD
NLRP3 knockouts are strongly protected from dopaminergic neuron loss and avoided α-synuclein pathology in PD mouse models, supporting NLRP3 as a therapeutic target [12, 42]. Table 2 highlights small molecule drugs that have been shown to inhibit inflammasome activation.
Summary of key inflammasome targeting therapeutics in development
Glibenclamide protects against paraquat and maneb exposure in mouse models. When treated with the inflammasome inhibitor, mice had decreased expression of NLRP3 as well as caspase-1 and IL1β relative to those mice treated only with the inflammasome activators [28]. BOT-4-ene, 3,4-methylenedioxy-b-nitrostyrene, and BAY 11-7082 act by similar mechanisms, interfering with the ATPase activity of NLRP3. BOT-4-ene acts through alkylation of NLRP3, encouraging ubiquitination [114], while BAY 11-7082 inhibit NLRP3 through direct inhibition of the ATPase [115]. 3,4-methylenedioxy-b-nitrostyrene prevents the formation of ASC specks [116]. Parthenolide, a herbal compound, inhibits NFκB through inhibition of caspase-1 protease activity [115]. JC-171 has been developed as a therapeutic for multiple sclerosis and has been shown to block IL1β through interruption of NLRP3 complex formation [117]. Fc11a-2 has been investigated as a therapeutic in models of IBS, acting by inhibition of the cleavage of the pro-form of caspase-1 [118]. Fenamates, a group of nonsteroidal anti-inflammatory drugs, have been showing promising results in AD models of NLRP3 inflammation, suppressing chloride efflux and IL1β levels to that of the wildtype [119]. OTL1177, another NLRP3 therapeutic that has been investigated for a potential protective effect in AD, resulted in increased cognitive function and decreased microglial activation [120].
Notably, MCC950 a highly selective NLRP3 inhibitor and is observed to bind exclusively to the NLRP3 inflammasome. In primary murine microglia, inflammasome assembly is halted after treatment with MCC950 after α-synuclein mediated inflammasome activation. In mice, MCC950 improves motor deficit symptoms and prevents α-synuclein accumulation and dopaminergic neuron death [12]. In the MPTP model, MCC950 treatment prevents microglial activation and NLRP3 activation [121]. Belnacasan is another small molecule drug that has progressed to the clinical stage, suppressing cytokine secretion and exhibiting anti-inflammatory features in rheumatoid arthritis [122]. Though having demonstrated success in animal models, these first generation inflammasome inhibitors have not yet progressed past early clinical studies. However, they remain highly promising therapeutics for disease modification in PD [123].
The use of natural products and lifestyle changes in order to inhibit inflammasome activation has also been explored [124]. Ellagic acid was shown to significantly reduce NLRP3 activation and ultimately DA neuronal death in MN9D and BV-2 cell lines primed with LPS [125]. Melatonin has been proposed to have a protective effect against inflammasome activation as well as being protective against neurotoxicity in dopaminergic neurons in an MPTP model. It halted microglia activation and prevented the secretion of IL1β [126]. There are intriguing links between sleep and circadian disruption which impact on inflammasome activation, but these have not been explored in PD [127]. Caffeine consumption, which is linked to a significantly lower risk of PD, has recently been shown to inhibit inflammasome activation in D-galactose treated rat models of neuroinflammation [128, 129]. In exercise-based studies, mice that were exposed to regular treadmill exercise after MPTP treatment exhibited decreased microglial activation and colocalization between NLRP3/Iba1 [130].
Given that inflammasome activation markers have been shown to be elevated in PD patient biofluids, they could have utility as markers of disease diagnosis or progression or to aid in clinical trial monitoring. However, antibodies associated with these inflammasome markers have been sub-par, generating unreliable results in terms of being able to differentiate the proteolytically active forms from the inactive pro-forms. Additionally, there is currently no way to determine if the elevated inflammasome markers observed in PD patients are a result of NLRP3 activation or if they are generated through other inflammasomes which could be relevant to PD [19].
CONCLUSIONS
A great deal of exciting progress has been made over the last 10 years in terms of identifying NLRP3 inflammasomes activation as a key driver of disease pathology in PD and demonstrating that it could be an important druggable target to slow or halt disease progression through multiple mechanisms. Misfolded α-synuclein, a hallmark of PD pathophysiology, has also emerged as the key inflammasome trigger in PD, both in the CNS and systemically. Despite the rapid progress that has been made, much remains to be understood in terms of the early inflammasome activation triggers in PD and the precise regulatory mechanisms that suppress inflammasome activation in the healthy brain. How and why inflammasome activation becomes persistent and unresolving in PD is another unanswered question, which remains challenging to address using current animal models. Although we have discussed several key triggers of inflammasome activation and downstream mechanisms that relate to dopaminergic degeneration, our mechanistic understanding of these processes is still quite limited, including what might be most relevant to human PD beyond α-synuclein aggregates. Further, research in inflammasome activation in PD has been focused primarily to investigate NLRP3, likely a result of its array of potential activators. This highlights a significant knowledge gap, as there remains much to be discovered for disease pathology in relation to other inflammasomes. Overall, much remains to be discovered in terms of inflammasome activation mechanisms and triggers during clinical PD, particularly at the gut-brain axis and beyond the CNS. In parallel, several inflammasome inhibitors targeted for CNS indications are being developed and some have entered early clinical trials. Outcomes from these trials will inform if inflammasome activation in the CNS could have therapeutic potential for disease modification in PD or if further refinements in therapeutic strategies would be required.
Footnotes
ACKNOWLEDGMENTS
RG was supported by an Advance Queensland Mid-Career Fellowship (AQRF03116-17RD2).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.