
Abstract
Alzheimer’s disease is a pervasive neurodegenerative disease that is estimated to represent approximately 70% of dementia cases worldwide, and the molecular complexity that has been highlighted remains poorly understood. The accumulation of extracellular amyloid-β (Aβ), intracellular neurofibrillary tangles formed by tau hyperphosphorylation, and neuroinflammation are the major pathological features of Alzheimer’s disease (AD). Over the years, there has been no apparent breakthrough in drug discovery based on the Aβ and tau hypotheses. Neuroinflammation has gradually become a hot spot in AD treatment research. As the primary cells of innate immunity in the central nervous system, microglia play a key role in neuroinflammation. Toll-like receptor 4 (TLR4) and nucleotide-binding oligomerization domain-like receptor 3 (NLRP3) inflammasomes are vital molecules in neuroinflammation. In the pathological context of AD, the complex interplay between TLR4 and the NLRP3 inflammasomes in microglia influences AD pathology via neuroinflammation. In this review, the effect of the activation and inhibition of TLR4 and NLRP3 in microglia on AD pathology, as well as the cross-talk between TLR4 and the NLRP3 inflammasome, and the influence of essential molecules in the relevant signaling pathway on AD pathology, were expounded. In addition, the feasibility of these factors in representing a potential treatment option for AD has been clarified.
INTRODUCTION
Alzheimer’s disease (AD) is currently the most common neurodegenerative disease among older individuals and is characterized by cognitive impairment and progressive memory loss. Current data suggest that the incidence of dementia is expected to double in Europe and to triple worldwide by 2050; moreover, the statistics would be even higher if cases were diagnosed solely by the biological definition of AD [1]. The pathology of AD is characterized by extracellular amyloid-β (Aβ) deposits forming senile plaques and intracellular tau protein hyperphosphorylation forming neurofibrillary tangles [2]. The increased levels of inflammatory cytokines and chemokines that are detected in the postmortem brains of both AD patients and amyloid precursor protein (APP) transgenic animals [3] suggests a significant role for neuroinflammation in the development of AD. The importance of persistent and excessive neuroinflammation in the AD progression merits further research.
Microglia are the most significant innate immune cells in the central nervous system (CNS) and can also be considered as macrophages of the CNS [4]. In a healthy brain, microglia are in a resting state and mainly play an immune surveillance role [5]. The inflammatory phenotype of microglia plays an indispensable role in neuroprotection. Nevertheless, persistent aberrant microglial activation and associated neuroinflammatory responses exacerbate the progression of AD and other neurodegenerative diseases [6–8]. Genetic studies on late-onset AD (LOAD) have also identified risk alleles that regulate innate immune activity, which is highly expressed in AD brain-resident macrophages and microglia [9, 10]. These findings fully illustrate the criticality roles of microglia and immune responses in the pathogenesis of late-onset AD, which requires further investigation.
Pattern recognition receptors (PRRs) stimulated by endogenous tissue injury or exogenous pathogen invasion can recognize abnormal proteins, pathogens, and cellular debris to initiate inflammatory reactions [11]. As a type of PRRs, Toll-like receptors (TLRs) can detect invading pathogens and tissue damage to initiate inflammatory responses; therefore, both exogenous pathogen-associated molecular patterns (PAMPs) and endogenous damage-associated molecular patterns (DAMPS) may be ligands for TLRs. Currently, 10 TLRs have been observed in humans, and 12 TLRs have been observed in mice. When TLRs are activated, a series of signaling cascades will be triggered, thus activating some transcription factors and thereby promoting the expression of some inflammatory factors, cytokines, chemokines, or growth factors [12, 13]. TLR4 can induce autoimmunity or immune tolerance after organism tissues or cells are stimulated by injury, hypoxia, stress, and other factors that are critically involved in multiple diseases such as arthritis, atherosclerosis, tumors, and systemic lupus erythematosus. Importantly, previous studies have shown that TLR4 functions in the brain and controls physiological and pathological processes, including AD [14].
The inflammasome is a multiprotein complex that is essential for innate immunity and is mainly distributed within the cytoplasm. Inflammasomes can be found in the cytoplasm of microglia, neurons, and astrocytes. Among the many inflammasomes, the NLRP3 inflammasome has been well studied. Its main structure consists of the NLRP3 sensor protein, the apoptotic protein associated with the adaptive protein (ASC) containing the caspase activation and recruitment domain (CARD), and the pro-caspase-1 effector protein [15, 16]. Moreover, stimulated microglia can intracellularly activate the NLRP3 inflammasome to mediate chronic neuroinflammatory responses and even lead to neuronal death and pyroptosis, thus promoting AD pathologic development [17, 18].
TLR4 can affect the pathological process of AD alone and promote the action of the NLRP3 inflammasome to further aggravate AD progression. Additionally, TLR4 activation can boost the production of NLRP3 and pro-IL-1β by activating the transcription factor NFκB, thus resulting in the disturbance of the synthesis of the NLRP3 inflammasome and the release of the inflammatory factor IL-1β. Most previous reviews have respectively focused on TLR4 or the NLRP3 inflammasome to illustrate the impact of related molecules on the occurrence and development of AD; however, they have failed to observe the two factors in tandem and have rarely focused on the role that they play in microglia [18–21]. When considering the new findings, this review summarizes the TLR4 and the NLRP3 inflammasome that mainly function in microglia in AD progression. In addition, we present relevant potential therapeutic targets and recent advancements in AD immunotherapy strategies.
THE INFLUENCE OF TLR4 IN ALZHEIMER’S DISEASE
The single nucleotide polymorphisms (SNPs) of TLR4 are related to LOAD, which is reflected in the fact that TLR4/Asp299Gly and TLR4/11,367 polymorphisms significantly increase the risk of LOAD in the Italian and Chinese populations, respectively [22, 23]. Compared to controls, the TLR4 expression was upregulated in the postmortem brains of patients with AD with excessive immunoreactivity in glial cells surrounding amyloid plaques [20]. These correlation findings suggest that TLR4 may play an important role in AD, and with a series of studies have found that TLR4 activation or inhibition is closely related to the occurrence and development of AD, although there is still some controversy concerning the results.
Activation of TLR4 in microglia in AD
In the pathological development of AD, TLR4 in microglia represents a double-edged sword, with both beneficial and detrimental effects [3]. There is sufficient evidence to demonstrate that the activation of TLR4 in microglia exerts Aβ scavenging and neuroprotective effects. LPS is the primary ligand of TLR4 and the main object of immune surveillance, which directly and specifically binds to TLR4 and activates the TLR4/NFκB signaling pathway. An earlier study found that Aβ in the culture medium was reduced by approximately half after microglia were treated with LPS in vitro. Tlr(Lps-d)/Tlr(Lps-d) mice failed to exhibit Aβ uptake after stimulation with a TLR4 ligand (but not with a TLR9 ligand) in vitro. This result suggests that the activation of TLR4 in microglia enhances the clearance of Aβ [24]. Moreover, microglia inhibit Aβ deposition and protect neurons from Aβ-mediated neurotoxicity after activation by the TLR4 signaling pathway in the early stages of Aβ amyloidosis. In vitro or in vivo experiments have shown that microglia treated with a low dose of TLR4 agonist (LPS/Monophosphoryl lipid A [MPL]) in advance in an Aβ oligomeric environment can reduce the production of the proinflammatory factor tumor necrosis factor-α (TNF-α) and can produce the neuroprotective factor interferon-β (INF-β) [25]. When microglia were treated with early low-dose TLR (TLR4/TLR2) ligands (MPL/Pam3Cys), the perception and clearance of soluble Aβ were promoted. This treatment exerted a protective effect on the memory and synaptic function of rats that were intravenously injected with Aβ1 - 42 [26]. The results of these two experiments indicate that the beneficial effect of microglial TLR4 receptor activation is closely related to the ligand concentration and time period of action. Interestingly, new findings suggest that CD14 and TLR4 can synergistically promote the internalization of fAβ42 by microglia and that this internalization requires the presence of clathrin [27]. Furthermore, this finding demonstrates that the activation of TLR4 in microglia to produce beneficial changes in AD pathology requires other molecular synergies. In summary, the activation of TLR4 in microglia has beneficial effects on AD but is influenced by the ligand dose and time period of action and requires other factors for coordination. Therefore, we need to identify more relevant influencing factors in future studies to better elucidate the appropriate mechanisms better and to identify targets for immunotherapy of AD.
However, related experimental results have also shown that the activation of TLR4 in microglia has deleterious effects on AD. For example, the supernatant of LPS-stimulated wild-type (WT) microglia was more likely to cause more extensive neuronal death than the supernatant from TLR4-defective microglia, thus suggesting that TLR4 plays an essential role in neurotoxic products released by microglia from the perspective of gene variants [20]. In earlier experiments, it was found that microglial activation and significantly increased amyloid deposition occurred in transgenic mice (APP 717 V-F) overexpressing mutant human APP and containing apolipoprotein E (ApoE) that is intracerebroventricularly administered with LPS for two weeks [28]. Due to the fact that LPS acts as a TLR4-related ligand, a subsequent study found that APPswe transgenic mice had large numbers of proliferating microglia and were activated in the cortex and hippocampus after 12 consecutive weeks of weekly intraperitoneal injections of LPS. However, Aβ in the brain was unexpectedly and significantly increased compared to control mice [29]. Moreover, LPS activates TLR4 in BV2 microglia, induces the release of the proinflammatory factor interleukin-1β (IL-1β) through the TLR4/MyD88/NFκB signaling pathway, and damages to PC12 neurons [30]. Genome-wide association studies have demonstrated that triggering receptors expressed on myeloid cell 2 (TREM2) are recognized as playing a protective role in AD pathogenesis [31, 32]. Additionally, there is evidence that LPS-induced activation of TLR4 in microglia may inhibit TREM2 expression, thereby exhibiting deleterious effects on AD [33]. Zhou et al. found that the expression of TLR4 appeared to be upregulated, whereas in contrast, the expression of TREM2 was significantly downregulated after APP/PS1 transgenic mice were given intraperitoneal LPS. Although the expression of Aβ was not significantly altered, it remarkably aggravated cognitive dysfunction and increased neuronal apoptosis in APP/PS1 transgenic mice [33]. Interestingly, inhibitors of JNK or NFκB, both of which constitutes parts of the MyD88-dependent pathway of TLR4 [34, 35], can restore the low expression of TERM2 caused by the activation of TLR4 in microglia by LPS [36]. These results confirm that the activation of TLR4 in microglia also has adverse effects on AD pathology, which is in marked contrast to previous results.
The above-mentioned results illustrate that TLR4 is a receptor in the microglia that induces inflammatory signaling pathways; moreover, whether the activation of TLR4 can be an immunotherapy target of AD remains a complex and contradictory issues that requires further clarification.
Inhibition of TLR4 in microglia in AD
Neuroinflammation is one of the essential pathologies of AD. Thus, the alleviation of neuroinflammation is an important target for treating AD [37, 38]. Several studies have confirmed that the inhibition of TLR4 in microglia reduces neuroinflammation and exerts neuroprotection. A novel polysaccharide (PTP70-2) isolated from Polygala tenuifolia can inhibit LPS-activated TLR4 in microglia through TLR4/MyD88/NFκB signaling to reduce neuroinflammation-induced neurotoxicity, thereby preventing neuronal death [39]. Harpagide, which is the main bioactive component of Scrophulariaceae, can reduce angiotensin II (Ang II)-mediated neuroinflammation, thus resulting in neurotoxicity and blood–brain barrier destructive effects by inhibiting the TLR4/MyD88/NFκB pathway [40]. Although the inhibition of the TLR4-mediated MyD88/NFκB signaling pathway is more common in regulating neuroinflammation, there is evidence that other signaling pathways can also be regulated to alleviate neuroinflammation [41–43]. For example, the novel polysaccharide ATP50-3, which is the purified crude polysaccharide AT50 from Acorus tatarinowii, was able to exert anti-neuroinflammatory and neuroprotective effects not only by both inhibiting the TLR4-mediated MyD88/NF-κ signaling pathway in microglia and the TLR4/PI3K/Akt signaling pathway [44]. The following experimental results further demonstrate that inhibition of TLR4 in microglia is anti-inflammatory and neuroprotective; in addition, it attenuates memory impairment and exerts other beneficial effects. In an AβO-mediated mouse model of AD, AβO-induced memory impairment was abrogated by selective TLR4 receptor antagonists; consistently, AβO failed to induce memory impairment and glial activation in TLR4 knockout mice [45]. Furthermore, DL0410 inhibited the activation of the TLR4/MyD88/TRAF6/NFκB signaling pathway in LPS-stimulated microglia, which acted against neuroinflammation and improved synaptic plasticity, and maintained the integrity of the blood–brain barrier. Significantly, the cognitive and learning functions of D-galactose-induced AD-like mice were considerably improved [46]. Ubiquitin-specific protease 8 (USP8) regulates the transition of microglia from a proinflammatory phenotype to an anti-inflammatory phenotype by inhibiting the TLR4/MyD88/NFκB signaling pathway in microglia and alleviates LPS-induced cognitive and motor deficits in mice [47]. In summary, these experimental results illustrate that the inhibition of TLR4 in microglia has beneficial effects on AD; thus, the above-mentioned compounds or extractions that inhibit the TLR4 pathway may be targets for treating AD (Table 1).
Overall, the current research results showed that microglia are mainly responsible for fragmental phagocytosis and inflammatory reactions. TLR4 is especially expressed on the surface of microglia in the CNS, and its central activation ligands include different aggregation forms of Aβ, LPS, and MPL. In the view of Fig. 1, the activation of TLR4 can play a positive role when considering Aβ pathology; however, from the perspective of inflammation, the inhibition of TLR can protect neurons.
The compounds or extractions targeting the inhibition of the TLR4 pathway in AD
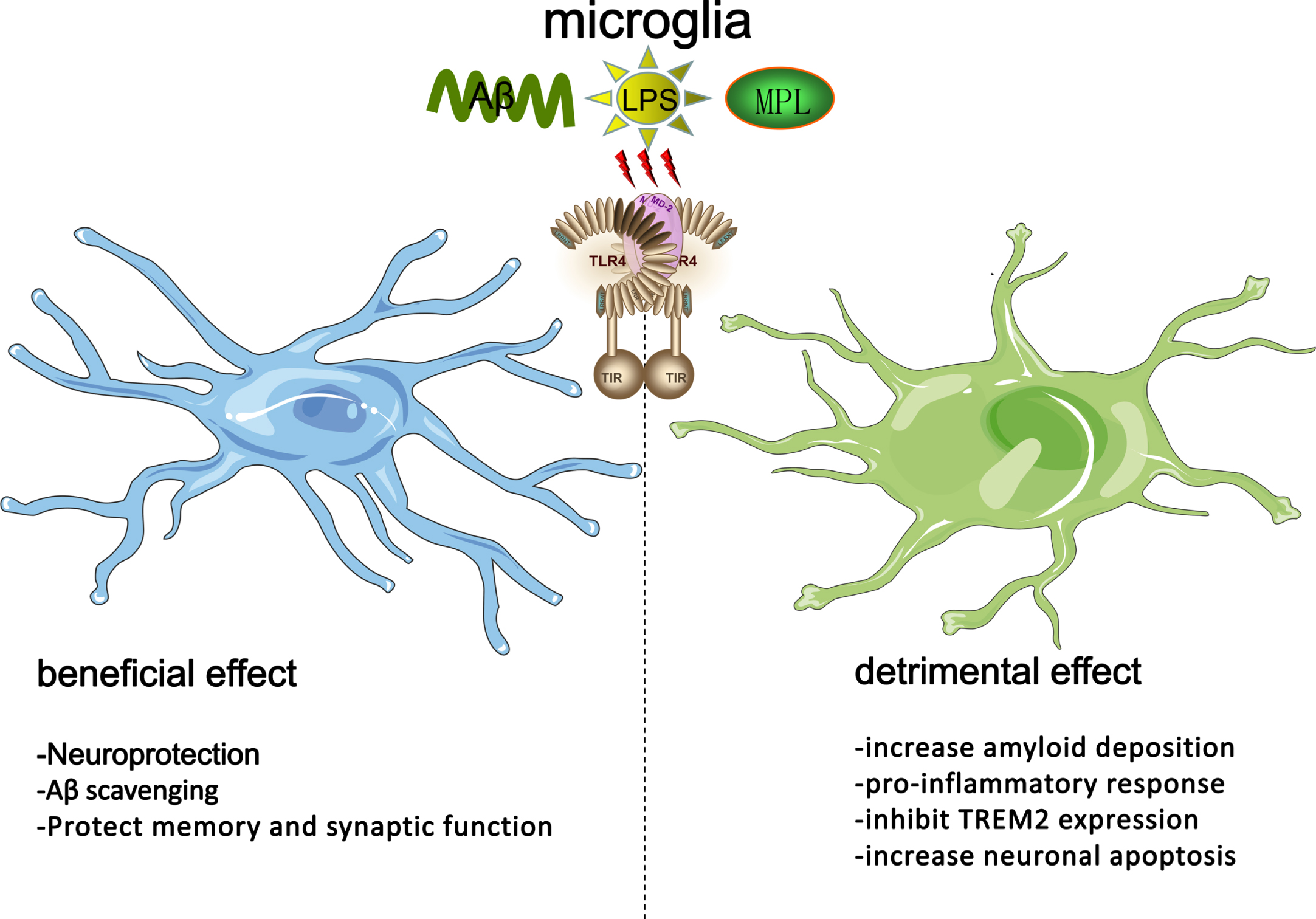
The role of TLR4 played in microglia and its ligand in AD. The beneficial effect of the activation of TLR4 in microglia via stimulation of LPS/MPL/Aβ oligomers/Aβ protofibrils includes neuroprotection, Aβ scavenging, and protection of memory, and synaptic function. However, this beneficial effect must depend not only on the ligand dose and time period of action but also on required coordination by other factors. The detrimental impact of the activation of TLR4 in microglia via stimulation of LPS/MPL/Aβ oligomers/Aβ protofibrils includes increasing amyloid deposition, the proinflammatory response, neuronal apoptosis, and inhibiting TREM2 expression.
THE INFLUENCE OF THE NLRP3 INFLAMMASOME IN ALZHEIMER’S DISEASE
Human genetic studies have found that the genetic variation in the 5’-flanking rs2027432 polymorphism of NLRP3 is related to LOAD with significantly increased susceptibility to LOAD in Northern Han Chinese individuals [48]. Clinical data have also shown that the NLRP3 inflammasome was overexpressed in the postmortem brains of AD patients [49]. The association of the NLRP3 inflammasome and AD has attracted extensive attention, and it is crucial to better understand the role that NLRP3 plays in AD, as well as the underlying mechanism by which NLRP3 functions in AD, which may provide new insights into AD progression and the potential possibility of using NLRP3 as a drug target for AD.
The influence of the NLRP3 inflammasome in microglia on Aβ pathology
As one of the DAMPs, the NLRP3 inflammasome of microglia can be activated by the different forms of Aβ, including Aβ oligomers and protofibrils [50]. However, Aβ can activate the NLRP3 inflammasome via different pathways. Aβ can be recognized not only by TLR4 across the cell membrane [51] but also by ASC specks of the NLRP3 inflammasome in the cytoplasm [52], after which inflammatory signals are transmitted to mediate neuroinflammation. Previous studies have found that Aβ activates NLRP3 in microglia in multiple ways. We summarize these methods of activation in Fig. 2. First, TLR4 mediates NLRP3 inflammasome activation in microglia stimulated by Aβ [51]. As a downstream target of TLR4/Myd88, NFκB can be activated, thus increasing the NLRP3 transcription. Conversely, with the increase in fibrillar Aβ in lysosomes in microglia, Aβ can induce cytoplasmic lysosome damage, which releases cathepsin B to endogenously activate NLRP3 [53]. Alternatively, Wang and colleagues found that Aβ-induced microglial activation and mitochondrial dysfunction, thus leading to reactive oxygen species (ROS) accumulation, followed by the activation of the NLRP3 inflammasome [54]. Feng and his colleagues further demonstrated that Aβ can activate the NLRP3 inflammasome in microglia through the TXNIP/TRX/NLRP3 pathway [55]. Some innovative research has additionally confirmed that interference with the TXNIP/NLRP3 pathway may inhibit the activation of NLRP3 activated by Aβ, which further conforms that Aβ can activate the pathway. Physical exercise can inhibit the increase in TXNIP and the activation of the NLRP3 inflammasome in the hippocampus induced by Aβ1-40 stimulation, which indicates that physical activity can inhibit the activation of the NLRP3 inflammasome by inhibiting the TXNIP/NLRP3 pathway [56]. Furthermore, resveratrol can also reduce the TXNIP/TRX/NLRP3 pathway activation induced by Aβ [55].
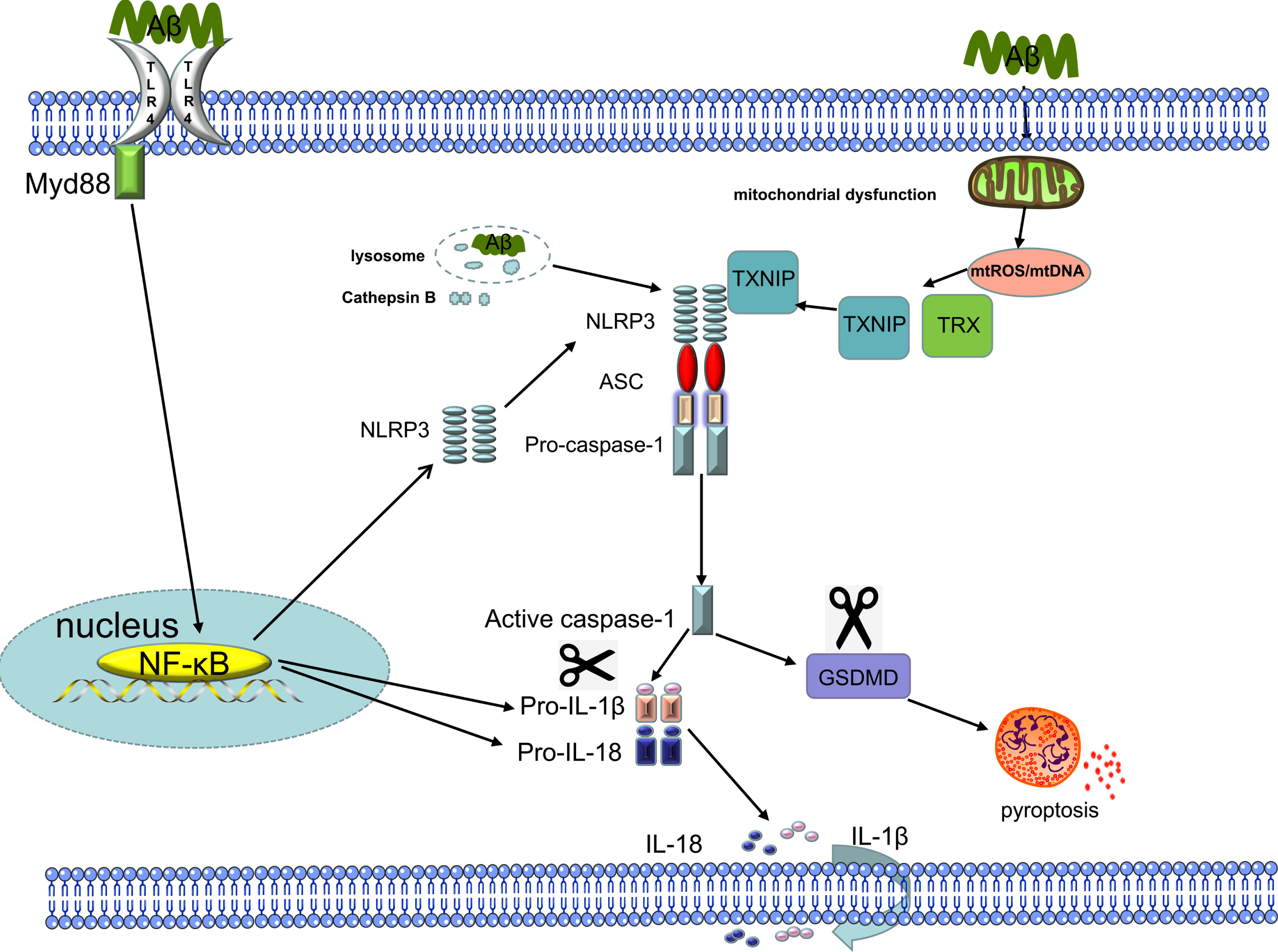
Mechanisms of Aβ activation of the NLRP3 inflammasome. Fibrillar Aβ, which is one of the PAMPs, induces the activation of NFκB through PRRs, including TLR4, to promote the expression of NLRP3 and pro-IL-1β. NLRP3, ASC, and pro-caspase-1 assemble to form the NLRP inflammasome to activate caspase-1, which not only cleaves pro-IL-1β to produce the proinflammatory factor IL-1β for extracellular secretion but also cleaves GSDMD to induce pyroptosis. Soluble Aβ damages the cytoplasmic lysosome to trigger the release of cathepsin B, which activates the NLRP3 inflammasome. Finally, Aβ oligomers induce mitochondrial dysfunction, which triggers ROS production and accumulation, mitochondrial DNA release, or cardiolipin externalization, thus activating the NLRP3 inflammasome.
Activation of the NLRP3 inflammasome can coordinate the removal of cell debris and promote tissue repair; however, it also encourages the release of the inflammatory factors IL-1β and IL-18 and induces pyroptosis. Therefore, when the NLRP3 inflammasome is normally activated by Aβ, a chronic inflammatory environment will be created, thus promoting AD occurrence and development [57]. The activation of NLRP3 in microglia may correspondingly affect Aβ further. An earlier study showed that the activation of the NLRP3 inflammasome in microglia induces polarization to the M1 state, which leads to increased Aβ deposition in APP/PS1 transgenic mice while exacerbating synaptic loss and cognitive impairment. Furthermore, in the APP/PS1/NLRP3–/– mouse model, the microglia were mainly of the M2 phenotype, thus reducing the extracellular Aβ load and protecting synaptic plasticity and cognitive ability [49]. Tejera and colleagues found that the activation of the NLRP3 inflammasome in microglia after intraperitoneal injection of LPS in aged AD transgenic mice decreased Aβ uptake and increased Aβ deposition. Interestingly, these pathological changes were not observed in young AD transgenic mice that received the same treatment [58], thus suggesting that NLRP3 may act as the medium by which peripheral inflammation influences the brain and that the inhibition of the NLRP3 can be as a novel therapeutic target for systemic infection. These studies confirm that NLRP3 inflammasome activation of microglia leads to increased Aβ deposition, thus adversely affecting AD. The inhibition of the NLRP3 inflammasome in microglia may be a feasible target for the treatment of AD; therefore, many studies have been performed concerning this point of view. Some researchers have found that under the action of MCC950 (which is an inhibitor of the NLRP3 inflammasome), microglia can enhance the phagocytosis of Aβ in vitro and reduce Aβ deposition in AD transgenic mice in vivo. Subsequently, Lonnemann et al. found that OLT1177, which is an NLRP3 inflammasome inhibitor, reduced cortical Aβ plaques and attenuated microglial activation to improve cognitive impairment and synaptic plasticity in APP/PS1 transgenic mice [60]. Similarly, a previous study found that the use of JC124 can inhibit the NLRP3 inflammasome, thus significantly reducing the activation of microglia and Aβ load while protecting neural plasticity and improving cognitivefunction [61].
Taken together, these experimental results illustrate the crucial role of Aβ in activating the NLRP3 inflammasome in microglia and briefly prove that the activation of the NLRP3 inflammasome is closely related to AD. The inhibition of NLRP3 in microglia for reducing Aβ pathology is a potential target for AD therapy. Thus, we need to further study the interaction of the NLRP3 inflammasome and AD pathology to develop effective therapeutic targets.
The interplay between microglia and the NLRP3 inflammasome in tau pathology
Tau hyperphosphorylation constituting extracellular neurofibrillary tangles is a significant pathogenesis of AD [62, 63]; however, studies on the interplay between the NLRP3 inflammasome in microglia and Tau are limited. Panda and his colleagues demonstrated that tau can activate the NLRP3 inflammasome of microglia to induce neuroinflammation [64]. Moreover, Stancu et al. found that tau and Aβ activate the NLRP3 inflammasome of microglia by a similar mechanism and also found that the inhibition of the NLRP3 inflammasome and the knockout of the ASC gene had beneficial effects on tau pathology in tau transgenic mice [65]. In recent years, a previous study has found that Tau22/Asc–/–and Tau22/Nlrp3–/–mice reduce tau phosphorylation by regulating tau phosphorylation-related kinases and phosphatases. this study also indicated that fibrillar Aβ can induce tau phosphorylation by activating the NLRP3 inflammasome in microglia [66]. A recent study also demonstrated that p-tau triggers the modulation of neuroinflammation and spatial memory in an NLRP3-dependent manner. Furthermore, p-tau was reduced in NLRP3 knockout mice [67].
In conclusion, tau can activate the microglial NLRP3 inflammasome, and the activated NLRP3 inflammasome can correspondingly hyperphosphorylate tau. This mechanism represents a vicious circle; if not treated in a timely manner, it will aggravate AD progression. Moreover, NLRP3 may be the bridge mediating the Aβ and tau pathology. The above-mentioned research has fully proven that the inhibition and deficiency of the NLRP3 inflammasome can reduce tau phosphorylation and cognitive dysfunction, which has beneficial effects on AD and is also a potential method for treating AD.
The interplay between microglia and the NLRP3 inflammasome in neuroinflammation
There is concrete evidence that neuroinflammation in AD is a chronic process that does not resolve on its own, and this inflammatory response can drive the development of the disease [68]. Microglia are the primary myeloid innate immune cells of the central nervous system [69]. Therefore, the neuroinflammation induced by the NLRP3 inflammasome in microglia plays an essential role in the occurrence and development of AD [57]. Some researchers have found that they were able to regulate neuroinflammation by inhibiting the activation of the NLRP3 inflammasome in microglia to alleviate AD progression. Specifically, Feng et al. found that intraperitoneal injection of dihydromyricetin (DHM) in APP/PS1 transgenic mice can reduce neuroinflammation by inhibiting the inflammasome of microglia, thereby promoting the clearance of Aβ and alleviating cognitive dysfunction [70]. Additionally, Li and colleagues found that pterostilbene alleviated Aβ1 - 42-induced neuroinflammatory responses in microglia by inhibiting the NLRP3/caspase-1 inflammasome pathway to reduce NO production and inhibit inducible nitric oxide synthase mRNA and protein expression [71]. Similarly, baicalin (BAI) attenuates neuroinflammation by inhibiting the NLRP3 inflammasome in microglia, thus ultimately reducing neuroinflammation-mediated neuronal apoptosis and improving cognitive function in APP/PS1 mice [72]. More recently, Zhang et al. found that AMS-17, which is a tertiary sulfonylurea compound, inhibited the expression of NLRP3 and its downstream components and cytokines in LPS-treated microglia to relieve neuroinflammation and inhibit the phagocytosis of microglia [73]. In addition to studies using compounds or extracts for treatment, there has also been a previous study using nanocarrier system-mediated gene therapy to assess the treatment of AD. This study found that polyethylene glycol-polyethyleneimine delivery of ROCK2-siRNA (PPSR) reduced neuroinflammation by inhibiting the Aβ-induced NLRP3/caspase-1 pathway in microglia [74]. Moreover, a recent study on microRNAs found that miR-138-5 reduced LPS-induced inflammatory responses in microglia by directly targeting the 3′-UTR of NLRP3. According to this mechanism, miR-138-5 also alleviated neuroinflammation and cognitive dysfunction induced by LPS in rats and reduced proinflammatory factors and microglial activation [75].
In summary, the regulation of the NLRP3 inflammasome in microglia can affect neuroinflammation, and the inhibition of NLRP3 activity in microglia via various methods can attenuate neuroinflammation, which demonstrates a way of using this mechanism to develop therapeutic strategies for AD is of great significance.
THE CROSSTALK BETWEEN TLR4 AND NLRP3 IN MICROGLIA
Three signaling pathways are generally believed to activate NLRP3 inflammation, including canonical NLRP3 inflammasome activation, noncanonical NLRP3 inflammasome activation, and alternative NLRP3 inflammasome activation. Among them, the crosstalk between TLR4 and the NLRP3 inflammasome can be observed in canonical NLRP3 inflammasome activation and alternative inflammasome activation [76, 77] (Fig. 3).
The role of TLR4 in canonical NLRP3 inflammasome activation in microglia
Canonical NLRP3 inflammasome activation is mainly divided into two steps: the priming step and the activation step. The activation of the NLRP3 inflammasome via TLR4 primarily occurs during the priming step, during which TLR4 binds to its ligand, then activates the transcription factor NFκB, and finally upregulates the expression of NLRP3, Pro-IL-1β, and Pro-IL-18 [78–80]. During the activation step, the pyrin domain (PYD) of the receptor protein NLR3 is linked to the adaptor protein ASC, whereas the CARD of ASC recruits pro-caspase-1 to activate caspase-1. The final activated caspase-1 can not only cleave the inactive proinflammatory cytokines pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18 but also lyse gasdermin D (GSDMD) to trigger pyroptosis [81, 82] (Fig. 3). This suggests that interfering with the interplay of TLR4 with the canonical NLRP3 inflammasome in microglia may be a potential target for treating AD. Zhong et al. found that epigallocatechin-3-gallate (EGCG) can alleviate neuroinflammation and exert neuroprotection by inhibiting TLR4/NFκB to reduce the activation of the canonical NLRP3 inflammasome in microglia [83]. Similarly, a previous study found that TLR4-IN-C34, which is a TLR4 inhibitor, alleviated neuroinflammation by inhibiting the TLR4/MyD88/NFκB/NLRP3 signaling pathway and reducing ROS generation in microglia [84]. Additionally, in a study of cerebrovascular disease, it was found that in permanent middle cerebral artery occlusion (pMCAO) rats, oral administration of agomelatine to inhibit the TLR4/NLRP3 signaling pathway in microglia alleviated neuroinflammation and pyroptosis [80]. There is evidence that microRNAs can affect canonical NLRP3 inflammasome activation. Specifically, Zhang and colleagues found that miR-124 inhibits the TLR4/MyD88/NFκB p65/NLRP3 signaling pathway in microglia to achieve anti-inflammatory effects [85]. In recent studies on neuroinflammation, it has been discovered that N-acetyldopamine dimer, and menstrual blood-derived endometrial stem cells can reduce the neuroinflammatory effects caused by lipopolysaccharide-stimulated BV-2 cells. This reduction occurs through the inhibition of the TLR4/NF-κB/NLRP3/Caspase-1 pathways [84, 87]. These findings suggest that the inhibition of the TLR4/NLRP3 signaling pathway in microglia reduces canonical NLRP3 inflammasome activation to attenuate neuroinflammation and enhance neuroprotection.
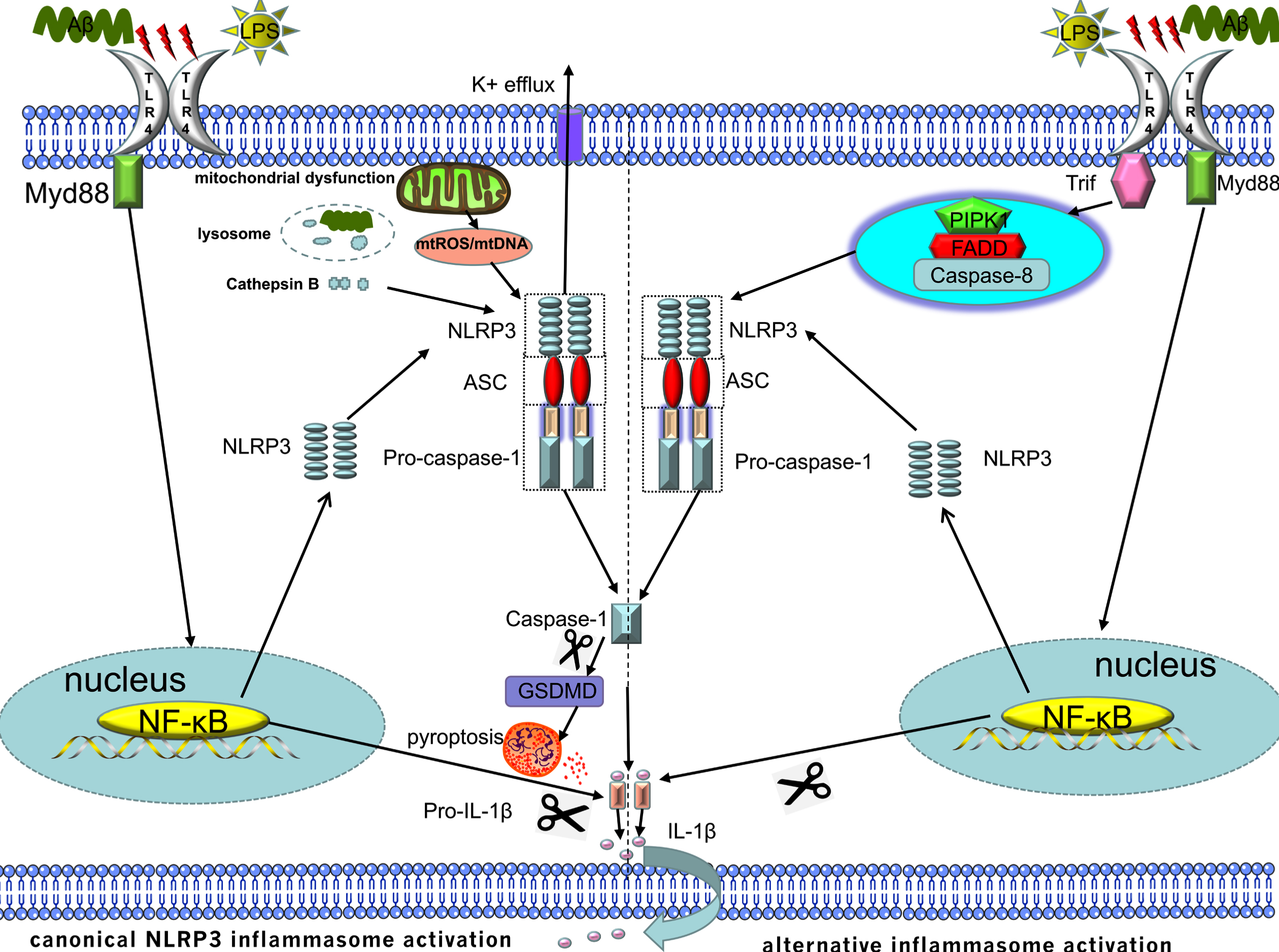
The crosstalk between TLR4 and NLRP3:1) Canonnical NLRP3 inflammasome activation is mainly divided into the priming and activation steps. TLR4 is primarily involved in the priming step of canonical NLRP3 inflammasome activation, during which TLR4 binds to ligands and promotes the expression of pro-IL-1β and NLRP3 through the TLR4/MyD88/NFκB signaling pathway. 2) Alternative NLRP3 inflammasome activation is just one step and only requires a single signal. The single TLR4 ligand can activate the NLRP3 inflammasome in monocytes via the TLR4/TRIF/RIPK1/FADD/CASP8 signaling pathway. However, this activation pathway has been limitedly studied in a limited manner in microglia and cannot induce K+ efflux, ASC speck formation, or pyroptosis.
The role of TLR4 in alternative inflammasome activation
Unlike canonical NLRP3 inflammasome activation, which requires two steps, alternative NLRP3 inflammasome activation requires only one step, which is also known as one-step NLRP3 inflammasome activation. The signaling pathway activated by the alternative NLRP3 inflammasome also differs from the canonical NLRP3 inflammasome activation. The former mechanism is activated by the TLR4/TRIF/RIPK1/FADD/CASP8 signaling pathway upstream of NLRP3, and the latter mechanism is activated via the TLR4/MyD88/NFκB signaling pathway. However, alternative NLRP3 inflammasome activation still requires NLRP3-ASC-caspase-1 signaling [88] (Fig. 3). Although the current research on the regulation of alternative NLRP3 inflammasome activation via the TLR4/TRIF/RIPK1/FADD/CASP8 signaling pathway in microglia is limited, it shows that research based on this mechanism has great potential.
The crosstalk between TLR4 and NLRP3 in other neurological conditions
In a previous study on stroke, it was discovered that agomelatine inhibits microglial activation by decreasing the expression of inflammasomes and reducing pyroptosis through the TLR4/NLRP3 pathway. As a result, it reduces neuroinflammation and ultimately mitigates brain injury in pMCAO rats [80]. Moreover, a previous study investigated the effects of atorvastatin, which is a traditionally used drug exhibiting a new mechanism, on cellular pyroptosis in the brains of mice with cerebral hemorrhage. The study results demonstrated that atorvastatin inhibits the TLR4/NLRP3 pathway, thereby preventing inflammasome activation in glial cells. This inhibition leads to a reduction in neuronal loss and ultimately protects against neurological deficits [89]. In a study investigating neurodegenerative diseases, the researchers discovered that Genkwanin has the ability to reduce neuroinflammation and neurotoxicity caused by MPP+ in a cellular model of Parkinson’s disease. This effect is achieved through the inhibition of the TLR4/NLRP3 inflammasome pathway, which is associated with the development of inflammation and cell pyroptosis [90]. The conventional anti-inflammatory drug dexamethasone attenuates brain damage due to subarachnoid hemorrhage by controlling microglial activation, which is in partially achieved through the inhibition of the TLR4/NLRP3 inflammasome pathway [91]. The aforementioned management options for other neurological disorders, which aim to alleviate neuroinflammation and protect neurons by inhibiting the TLR4/NLRP3 pathway, may also hold potential as treatment options for AD.
Potential AD therapeutic targets of IL-1 and NFκB
Based on the canonical NLRP3 inflammasome activation pathway, researchers have conducted further studies on essential molecules in the signaling pathway as potential targets. IL-1β, which is a downstream product of NLRP3 inflammasome activation, is an inflammatory factor that directly acts on target cells and is an attractive target for alleviating neuroinflammation and treating AD [92–94]. Additionally, He and colleagues found that the long-term inhibition of IL-1 signaling in 3xTg-AD mice alleviated AD-related pathologies, including Aβ pathology, tau pathology, and neuroinflammation [95]. Subsequent studies have found that anakinra, which is an IL-1 receptor antagonist, can not only improve synaptic plasticity in transgenic rats overexpressing AβPP but also alleviate the effect of AβOs on proteins involved in mitochondrial dynamics to attenuate synapse loss and cognitive dysfunction [96, 97]. However, some previous studies have found that inhibiting IL-1 also adversely affects AD. Wang et al. found that although IL-1R deficiency alleviated cognitive dysfunction in AD mice, it also reduced microglia phagocytic ability [98]. Similarly, a study found that the overexpression of IL-1β early in amyloid pathogenesis altered microglial gene expression profiles and promoted microglial growth to enhance Aβ plaque clearance [99]. As a significant downstream transcription factor in the TLR4 signaling pathway, NFκB promotes the expression of inflammatory mediators [100] and participates in the activation of the NLRP3 inflammasome [78]. When neuroinflammation induced by the activation of NFκB in microglia has a deleterious effect on AD [101], the regulation of NFκB is also an important target for treating AD. Kong et al. found that forsythoside B (FTS\bulletB) regulates NFκB signaling in APP/PS1 mice to attenuate neuroinflammation and cognitive impairment and to alleviate microglia-mediated neurotoxicity [102]. Moreover, Kim and colleagues found that the inhibition of the NFκB signaling pathway by isoorientin can reduce Aβ25 - 35-induced neuroinflammation and apoptosis in microglia [103]. A previous study also found that in LPS-treated microglia, Trichosanthes semen can activate HO-1 to inhibit NFκB signaling, thereby reducing neuroinflammation [104]. Although the NLRP3 inflammasome produces IL-1β and TLR4 activates NFκB, their respective downstream products can have a relevant effect on AD, thus indicating their importance in AD immunotherapy.
In summary, the regulation of IL-1 and NFκB can attenuate neuroinflammation and play a neuroprotective role, which is a good target for treating AD. However, there is evidence that the regulation of IL-1 and NFκB may affect the phagocytic function of microglia; Moreover, the question of how to maintain the balance between the two effects is a problem that we need to solve in the future.
CONCLUSIONS AND PERSPECTIVES
As PRRs, TLR4 and NLRP3 can sense the stimulation of endogenous tissue damage or the invasion of exogenous pathogens, thus triggering an inflammatory response to play a central role in immune protection. The Aβ or tau pathology of AD can excessively or persistently activate TLR4 and NLRP3 in microglia to generate chronic neuroinflammation in the brain, thereby promoting AD progression. Correspondingly, the activation of TLR4 and NLRP3 in microglia can also have adverse effects on Aβ or tau. TLR4 is involved in the priming step in canonical NLRP3 inflammasome activation and directly participates in the activation step in alternative NLRP3 inflammasome activation. Studies have shown that the inhibition of TLR4 or NLRP3 in microglia may attenuate Aβ or tau and even reduce inflammation and alleviate cognitive dysfunction. Some studies have also found that intervening molecules in the signaling pathway associated with TLR4 and NLRP3 can also alleviate AD-related pathological progression. Therefore, the regulation of the functions of TLR4 and NLRP3 in microglia may be a potential target for treating AD. However, many problems require further investigation. First, the inflammatory response is supposed to have a bidirectional functional effect on the brain; thus, the investigation of the appropriate time window for intervention to control the inflammation is a problem. Second, due to the fact that immunotherapy constitutes a double-edged sword, further research is needed to explore the dosage and administration of target drugs, as well as the adverse effects of modulating relevant targets.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
This work was supported by the Natural Science Foundation of Guangdong Province (2022A1515010593).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.