
Abstract
Inflammation and immune dysregulation have been linked to the pathogenesis and progression of Parkinson’s disease (PD), and represent an attractive target for therapeutic intervention, given the potential for repurposing of existing anti-inflammatory and immunomodulatory agents. Despite the fact that initial studies of drugs with secondary anti-inflammatory effects did not yield positive results, agents specifically targeting immune and inflammatory pathways may hold more promise. This article will briefly review the evidence base for targeting the immune system and neuroinflammation in PD, and discuss in detail the recently completed and currently active trials of primary anti-inflammatory/immunomodulatory drugs in PD.
Plain Language Summary
Parkinson’s disease is caused by a loss of dopamine-producing nerve cells in the brain. Recent research has suggested that activation of the immune system, leading to inflammation in the brain and body, can contribute to this loss. Current medications that are used to treat Parkinson’s disease only help with the symptoms, and do not slow down the damage to nerve cells in the brain. New treatments, aiming to reduce inflammation and thereby slow disease progression, are under investigation in a number of clinical trials which are reviewed in this article. These treatments include medications that have been used in other diseases, as well as new drugs designed to target inflammation and immune activation in PD. Some of these early studies have had encouraging results but further larger trials are needed to determine whether medications targeting inflammation will have benefit for people with PD.
INTRODUCTION
In spite of effective symptomatic therapies which improve many of the motor symptoms of PD, the neurodegenerative pathology inexorably progresses, such that two thirds of patients will develop significant postural instability and nearly half will develop dementia within the first 10 years, 1 with no treatments available to slow this process. A wealth of research has suggested that sustained neuroinflammation, driven by both the innate and adaptive immune system, can precede and perpetuate neuronal degeneration in PD, making it a potential disease modifying therapeutic target.2–4
McGeer et al. initially proposed that sustained neuroinflammation is linked to the development and progression of PD in 1988, demonstrating an increase in reactive HLA-DR+ microglia in the substantia nigra at post-mortem, which correlated with neurodegeneration. 5 Subsequent in vivo PET imaging studies have corroborated these findings using radioligands binding to translocator protein (TSPO), showing an upregulation of activated microglia in PD in disease-relevant sites. 6 Furthermore, microglial activation has been shown to correlate with motor symptom severity, 7 cognitive dysfunction 8 and dementia risk 9 in PD.
Microglia constitute the first line of the brain’s immune defense, responding to cell damage and pathogen-derived stimuli. Activation leads to the clearance of cellular debris and toxic proteins, subsequently leading to tissue repair. 10 Chronic activation of microglia leads to sustained secretion of pro-inflammatory cytokines, chemokines and neurotoxic factors, creating an uncontrolled inflammatory environment which is proposed to contribute to neurodegeneration in PD. Neuronal death, with associated release of misfolded alpha synuclein and other toxic species may lead to a vicious cycle of microglial activation andneurodegeneration. 11
The nucleotide-binding domain, leucine-rich–containing family, pyrin domain–containing-3 (NLRP3) inflammasome is a complex of proteins which forms on activation of microglia and other innate immune cells, and is a key amplifier of the inflammatory response. It has recently emerged as a potential contributor to the pathogenesis of PD, evidenced by elevated levels of NLRP3 and downstream cytokines such as IL1β in the blood in PD patients, 12 coupled with increased NLRP3 protein levels in dopaminergic neurons in the midbrain at postmortem. 13 NLRP3 inflammasome activation can be driven by the recognition and internalization of α-synuclein by microglia,14,15, 14,15 as well as by mitochondrial dysfunction, which is prominent in PD. 16 Upon inflammasome activation, pro-inflammatory cytokines IL-1β and IL-18 are released, and cell death can occur via pyroptosis. 16 The exact mechanisms of neurodegeneration due to inflammasome activation remain unknown; however, direct neurotoxicity and the seeding of protein aggregatesis proposed. 16
This pro-inflammatory environment in PD extends beyond the central nervous system. Multiple studies have documented elevated serum levels of inflammatory cytokines such as IL-6, TNF-α, IL-1β, IL-2, and IL-10 in PD, with a more inflammatory cytokine profile in early disease being associated with accelerated motor and cognitive decline.17,18, 17,18 An increase in the neutrophil to lymphocyte ratio (NLR), a marker of peripheral inflammation, can pre-date the development of PD and correlates with motor disease severity and dopamine transporter binding in the striatum.19–21 Moreover, numerous studies have revealed shifts in both innate and adaptive peripheral immune populations, with elevated levels of pro-inflammatory subsets. In the T lymphocyte population, this includes an increase in TH1 and TH17 pro-inflammatory effector cells, 22 and a lack of the CD8 lymphocyte immunosenescence that is normally seen with aging.23,24, 23,24 T cell subsets have been shown to correlate with motor severity25–27 and notably these effector T cells recognize alpha synuclein in PD, 28 and are present years before disease onset. 29 Studies have also demonstrated that there are fewer anti-inflammatory T-regulatory cells in PD,22,30,31, 22,30,31 and that these cells are less able to suppress the pro-inflammatory action of effector cells. 27 A similar pattern of a skew towards pro-inflammatory subsets is seen in other cell populations in the blood, including B cells,30,32, 30,32 NK cells33,34, 33,34 andmonocytes.35,36, 35,36
The relevance of the peripheral immune system in PD pathology has been explored in numerous PD models. In an in vitro model system using iPSC derived neurons from PD patients, neuronal death was promoted by autologous T lymphocytes and mediated by IL-17 signalling. 37 In vivo studies in animal models of PD have demonstrated that both innate and adaptive immune cells infiltrate to sites of a-synuclein pathology and attenuation of this infiltration leads to neuroprotective effects.38–40 In human PD, there is also growing evidence that peripheral immune cells can interact directly with central neuropathology. In postmortem PD brain, T lymphocyte infiltration has been reported in the substantia nigra in close proximity to dopaminergic neurons,5,39, 5,39 with greater T cell infiltration in PD withdementia. 9
Genetic and epidemiological studies provide added support for the role of the immune system in the pathogenesis of PD. Genome wide association studies demonstrate a link between PD risk and polymorphisms in the human leukocyte antigen locus as well as genes involved in antigen presentation and recognition, inflammation and the complement system.41,42, 41,42 Moreover, proteins implicated in genetic forms of PD, such as LRRK2, are highly expressed in cells of the innate immune system and are involved in numerous immune signaling pathways. 43 Patients with a diagnosis of an autoimmune disease, including inflammatory bowel disease, have an increased risk of developing PD, 44 and this risk is attenuated in patients on anti-TNF immunological therapy. 45 Use of non-steroidal anti-inflammatory drugs and immunosuppressive therapies has also been reported to be associated with a lower risk of PD in large epidemiological studies.46–48
Overall, there is compelling evidence that the immune system plays a pivotal role in the pathogenesis of PD (Fig. 1). Coupled with promising results from therapies targeting inflammation in pre-clinical models of disease, this suggests that modulating inflammation may be a highly promising disease modifying therapeutic approach.
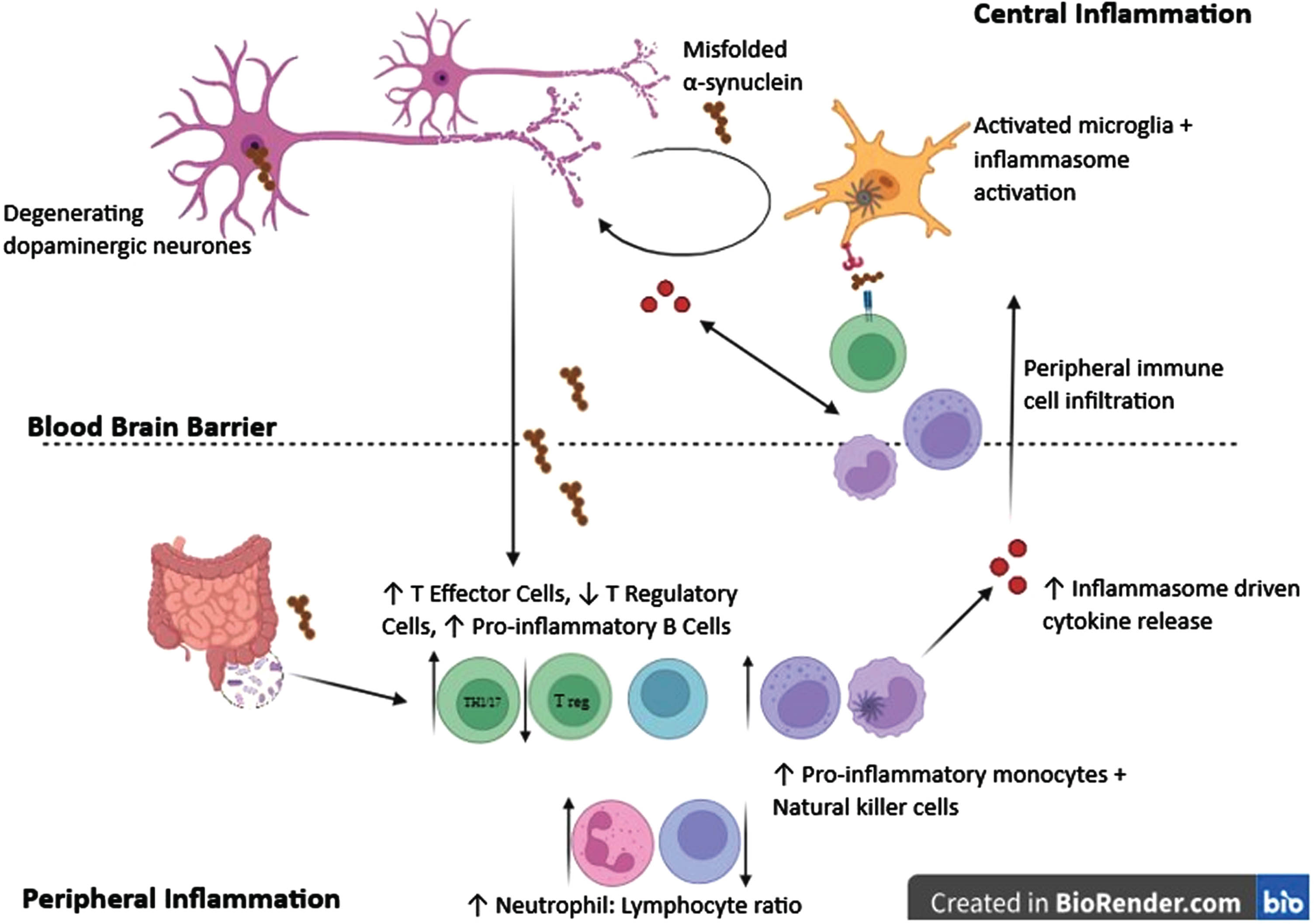
Overview of the immune system and inflammation in Parkinson’s disease (PD). Immune changes and inflammation occur both within the brain and periphery in PD. Activated microglia are found throughout the brain with evidence of NLRP3 inflammasome activation. This leads to the release of inflammatory cytokines which have a toxic effect on neurons, and release of chemokines which recruit peripheral immune cells to the central nervous system. Peripherally there is evidence of both innate and adaptive immune cell dysfunction with an increase in the neutrophil to lymphocyte ratio in the blood, and a shift towards a more inflammatory phenotype of T and B lymphocytes as well as natural killer cells and monocytes. NLRP3 inflammasome activation occurs within peripheral innate immune cells as well as centrally, and proinflammatory cytokines in peripheral blood are increased. Misfolded alpha synuclein may act as a driver of innate immune activation both peripherally and centrally, and trigger a specific adaptive immune response.
OVERVIEW OF ANTI-INFLAMMATORY/IMMUNOMODULATORY DRUGS IN PD CLINICAL TRIALS
Trials involving medications with secondary anti-inflammatory properties
Despite growing evidence for the role of the immune system in PD since the late 1980 s, historically there have been relatively few trials primarily targeting inflammatory mechanisms.
Trials using repurposed medications or supplements with secondary anti-inflammatory actions started to enter clinical trials in PD in the early 2000s.
Minocyline, a tetracycline antibiotic, was the first anti-inflammatory therapeutic agent tested in PD. It has been shown to penetrate the central nervous system and reduce microglial activation and proinflammatory cytokine production in animal models,49,50, 49,50 which is thought to be mediated through a reduction in the pro-inflammatory enzymes inducible nitric oxide synthase (iNOS) and caspase 1.50,51, 50,51 A phase 2 double blind placebo-controlled futility trial was completed in 2003 recruiting 200 participants randomized to receive either creatine, minocycline or placebo.52,53, 52,53 Futility criteria were a 30% reduction in Unified Parkinson’s disease rating scale (UPDRS) progression over the treatment period, based on data gathered from the placebo/tocopherol arm of the DATATOP (Deprenyl And Tocopherol Antioxidative Therapy Of Parkinsonism) trial. 54 Although minocycline was not rejected as futile, it was not progressed to phase 3, likely due to high discontinuation rates, safety concerns, and a lack of efficacy in slowing disease progression, as evidenced by comparable UPDRS scores to the placebo arm.52,53, 52,53
Pioglitazone, a peroxisome proliferator-activated receptor γ agonist, has been shown to reduce microglial activation and neuroinflammation in preclinical testing in animal models of PD.55,56, 55,56 It was taken forward to a phase 2 futility trial recruiting 210 participants, but there was no improvement in UPDRS motor scores or changes in the inflammatory cytokine IL-6 following 44 weeks of treatment.57,58, 57,58 Another diabetic medication, exenatide, a GLP-1 agonist, has demonstrated anti-inflammatory effects in animal models of PD.59,60, 59,60 It has also shown promise in a phase 2 randomized control trial of 62 participants: 48 weeks of treatment led to a significant beneficial effect on motor performance (UPDRS-III) in the treatment group. 61 A phase III trial of exenatide for 96 weeks in 200 participants (Exenatide-PD3) is now in its final stages. 62 Lixisenatide, another GLP-1 agonist has completed a phase 2 randomized placebo controlled trial in 156 participants with early PD, with a statistically significant reduction in motor progression in the treatment arm in comparison to placebo, suggesting the GLP-1 pathway may be a promising target in PD. 63
Statins are thought to exert numerous neuroprotective actions, including reducing alpha synuclein aggregation, oxidative stress and microglial activation. 64 Nevertheless, the PD-STAT trial, a large multicenter phase 2 study recruiting 235 participants, with a treatment duration of 2 years, demonstrated no beneficial impact of simvastatin on PD progression. 65 Similarly, a 48-week trial of lovastatin in 77 PD patients, showed a trend in reduction of UPDRS part III scores which did not reach statistical significance. 66 Inflammatory biomarkers were not assessed as a part of either of these trials.
Gemfibrozil (FHL-301), currently used in lipid modification, has also entered phase 2 clinical trials in PD (NCT05931484). Gemfibrozil has been found to increase the production of glial derived neurotrophic factor in astrocytes and protect dopaminergic neurons in PD models. 67 It is also thought to have anti-inflammatory effects mediated via a reduction in pro-inflammatory cytokines leading to lower production of nitrous oxide and expression of iNOS, 68 as well as reducing microglial activation and reducing release of pro-inflammatory cytokines in response to lipopolysaccharides. 69 Forest Hills Labs are running a phase 2, double blind placebo-controlled trial with a 3-week dose titration period followed by a 48 week treatment period in 32 participants with early phase PD, with change in MDS-UPDRS part III as the primary outcome measure. However, the trial does not appear to include analysis of inflammatory biomarkers. Completion is anticipated at theend of 2024.
Vinpocetine, an agent that improves cerebral blood flow and has anti-inflammatory effects through phosphodiesterase mediated pathways, was explored in a short, randomized, double-blinded study of 89 participants who received 2 weeks of treatment. 70 The active treatment arm showed reduced TLR2 and TLR4 gene expression and downstream production of the pro-inflammatory cytokine TNFα. 70 Cognitive performance also improved post treatment, although this finding is difficult to interpret in such a short study and likely due to a learning effect of repetitive testing over 2 weeks, evidenced by a similar increase in the standard therapy arm. 70 Nevertheless, the biological effect is of interest given that TLRs have been implicated in mediating an inflammatory response to aberrant alpha synuclein in PD, 71 and further exploration through a longer duration placebo-controlled trial could beconsidered. 72
Niacin, a form of vitamin B3, reduces the translocation of NFkβ, a key transcription factor in the inflammatory cascade, and the production of downstream inflammatory cytokines, in response to LPS stimulation in a cellular model of PD. 73 This is thought to be mediated through its interaction with the niacin receptor GPR109A. 73 Supplementation has minimal side effects hence this is an attractive therapeutic candidate. However, in a recent trial in 47 PD patients, no significant change in UPDRS-III scores was observed during the 6-month double-blind placebo-controlled phase. An improvement in motor function was observed in a subsequent 6-month open label phase, but a placebo response seems the likely explanation for this given the lack of benefit in the placebo-controlledphase. 74
Sulforaphane, a compound found in vegetables such as broccoli and cauliflower, has anti-oxidant, anti-inflammatory, and anti-apoptotic properties, which has sparked interest into its use as a supplement in numerous neurodegenerative disorders including PD. 75 In in vivo models of PD, sulforaphane reduced astrogliosis, microgliosis and pro-inflammatory cytokines IL-6 and TNF-α via nuclear factor erythroid 2 related factor 2 (Nrf2) mediated pathways. 76 A randomized placebo-controlled trial is currently underway, recruiting 100 people with PD and an MMSE score of≤27 and treating them with 6 months of supplements with primary outcome measures of cognitive change. Secondary outcome measures include changes in motor function and safety and tolerability of the supplement (NCT05084365).
Currently active trials targeting the immune system/inflammation
The growing awareness of the role of inflammation and the immune system in the etiology of PD has led to an increasing number of early phase trials of novel and repurposed drugs specifically targeting immune and inflammatory pathways. For this review, both ClinicalTrials.gov and WHO clinical trials registries were searched for all active interventional clinical trials of drugs with primary anti-inflammatory/immune modulating mechanisms in Parkinson’s disease. Eight clinical trials were identified as either active or recently completed (within the last two years) (Table 1).
Clinical trials of immunosuppressive/anti-inflammatory drugs in PD which are either active or recently completed
A phase 2 trial of azathioprine commenced recruitment in 2021 and is the first to repurpose a peripherally-acting immunosuppressant drug for PD. It is a randomized, double-blind, placebo-controlled trial of 60 patients with early PD, receiving treatment for one year, with a primary outcome measure of the gait-axial subscore of the MDS-UPDRS. 77 The results of this trial are expected in 2024 and remain highly anticipated as the drug itself does not cross the blood brain barrier. Hence if the results of this trial are positive, it will support the role of interplay between the peripheral and central immune system contributing to the progression of PD. The trial incorporates proof of mechanism outcome measures including immunophenotyping in the blood and cerebrospinal fluid and TSPO PET/MR imaging to evaluate brain inflammation. 77
Sargramostim, a human recombinant granulocyte-macrophage colony-stimulating factor (GMCSF) that can induce T-regulatory immune cell responses, is currently undergoing extended phase 1 testing. Initial results with 8 weeks of treatment in 20 patients with PD demonstrated target engagement in terms of increasing the numbers and suppressive function of peripheral T regulatory cells, and there was a modest reduction in UPDRS part III scores in the sargramostim-treated versus the placebo arm. 78 Unfortunately, there were numerous side effects at the 6 μg/kg/day dose, with daily subcutaneous injections leading to significant injection site reactions. 78 Ongoing multi-year trials with fewer participants receiving lower treatment doses (3 μg/kg on five days per week) are underway in a bid to improve its tolerability in the PD population, however its efficacy at this dose needs furtherexploration.79,80, 79,80
With concerns over the tolerability of Sargramostim, a novel, longer acting GMCSF, PDM608, is under development in a phase 1 safety, tolerability and dose finding study in healthy volunteers (NCT05950906). Following a single administration in MPTP mouse models, PDM608 has shown an anti-inflammatory effect with a reduction in activated microglia and increased T-regulatory cell numbers and function for 10 days. 81 If successful in humans, a single subcutaneous injection could be given 1-2 weekly, alleviating the need for daily injections, hopefully mitigating some of the adverse events seen in the Sargramostim trialsto date.
Montelukast, a cysteinyl leukotriene receptor type 1 (CysLT1) antagonist, currently licensed as an oral therapy for asthma, has shown efficacy in animal models of PD.82,83, 82,83 In a phase II open label trial in 15 patients, the medication was well tolerated with no significant safety concerns despite the high doses used (four times the licensed dose in asthma), with a significant improvement in MDS-UPDRS part I–III scores post treatment (EUCTR2020-000148-76-SE). Intelgenx has recently won approval from the Swedish Medicines Agency for a new formulation of montelukast, which is a thin film applied orally which can be absorbed directly and without water, and will be trialed in a large phase 2, multi-center, double-blind, randomized, placebo- controlled trial (EudraCT number 2023-504278-39-00) in PD. 84 This has a 50% higher bioavailability, bypasses first pass metabolism, thereby lowering potential toxic effects. 84 The new trial aims to recruit 90 participants who will be randomized to receive montelukast or placebo for a duration of 18 months.
With inflammation now implicated in a broad range of diverse diseases, research is focusing on identifying and modifying novel therapeutic targets. To date, two novel anti-inflammatory agents have completed trials in PD. NE3107 is an orally administered brain penetrant molecule that binds to and inhibits extracellular signal-regulated kinase (ERK) mediated pathways in macrophages, leading to a decrease in the downstream inflammatory cascade. 85 In a phase 2 double-blind placebo-controlled trial in 46 participants over 28 days, treatment with NE3107 resulted in a clinically meaningful benefit in PD with a 3-point reduction in UPDRS part III scores and a reduction in OFF time in comparison to placebo. 86 AKST4290 is a small molecule antagonist of CCR3, a chemokine receptor expressed on leucocytes, as well as on microglia and astrocytes in neuroinflammatory conditions, 87 which plays a role in T cell migration into the brain. 88 In a randomized placebo-controlled phase 2 trial with 12 weeks of oral treatment in 110 participants with PD, there was no evidence of a beneficial change in the primary outcome measure of motor progression (MDS-UPDRS Part III) (NCT04369430). 89
The NLRP3 inflammasome, an integral component of the innate immune system, has attracted much interest in recent years, leading to the development of multiple novel selective inhibitory molecules. Some of these molecules can cross the blood-brain barrier, with the potential for translation to PD clinical trials. Nodthera have recently announced completion of their phase 1b/2a trial on NT-0796, an orally administered brain penetrant NLRP3 inhibitor. 90 Full details of the trial protocol and results are not yet available, but a press release has suggested that the novel molecule had an impact on peripheral and central cytokine production, lowering levels of IL-1β, IL-6, CCL2, CXCL1, and CXCL8 in up to 10 PD patients following 28 days of treatment, with minimal adverse events. 90 A phase 1b, randomized placebo-controlled trial investigating selnoflast, another orally administered NLRP3 inflammasome inhibitor, is underway. The investigators are aiming to recruit 72 participants with early PD, treating participants for 28 days with either selnoflast or placebo, with primary outcome measures related to safety and tolerability and secondary outcomes assessing pharmacokinetics and target engagement using [ 18 F]-DPA-714 TSPO PET. Results are expected in 2025 (NCT05924243).
The authors of this review are leading a phase 2 randomized, double-blind, placebo-controlled trial using the NLRP3 inflammasome inhibitor dapansutrile 91 , developed by Olatec Therapeutics. The trial is currently in set-up. Dapansutrile penetrates the blood-brain barrier and has neuroprotective effects in MPTP mouse models of PD. 92 Early phase trials in healthy volunteers, 93 gout, 94 and heart failure 95 have shown a good safety and tolerability profile with a treatment-induced reduction in levels of pro-inflammatory cytokines IL1β and IL6 in the blood. This trial aims to recruit 36 participants with early phase PD, with an initial randomized double-blind placebo-controlled phase of six months, followed by a 6-month open label phase. 91 The primary outcomes will be safety and tolerability, with blood and CSF inflammatory markers and [ 18 F]-DPA-714 TSPO PET imaging used to evaluate target engagement, and exploratory clinicalmeasures.
Two other early phase trials of novel NLRP3 inhibitors in PD have recently been announced. Ventyx Biosciences have completed a phase 1 trial in healthy volunteers with their novel CNS penetrant NLRP3 inflammasome inhibitor VTX3232, which was well tolerated with only mild to moderate adverse events recorded and positive results noted in whole blood IL1β stimulation assays. 96 They have announced a phase 2a trial in patients with PD, however there is no further information available about trial design yet. 96 Similarly, Zydus have completed a phase 1 trial in which 30 healthy male volunteers were recruited into a single ascending dose study, receiving a single dose of ZYIL1, and 18 healthy male volunteers were recruited into a multiple ascending dose study receiving ZYIL1 for a total of 14 days. A total of 12 adverse events were recorded over the two studies ranging in severity from mild to severe, and there was evidence of >90% reduction in IL1β and >70% reduction in IL18 release in whole blood from treated participants stimulated with LPS/ATP to activate the NLRP3 inflammasome in comparison to pre-treatment levels. 97 They have recently gained FDA approval to start a phase 2 clinical trial in PD but further details are currently unavailable. 98 NLPR3 inflammasome inhibitor studies which have recently completed, are active or planned, are summarized in Table 2.
Clinical trials of NLRP3 inflammasome inhibitors in PD
TBC, To be confirmed.
CONCLUSION
There is considerable evidence that immune activation contributes to the pathogenesis and progression of PD. Multiple tractable therapeutic targets within immune and inflammatory pathways have been identified and whilst as yet no definitively positive results have emerged from large-scale trials, several early phase studies have reported promising efficacy signals, warranting further exploration.
Immune and inflammatory mechanisms interact with multiple other complex pathological processes involved in the pathogenesis of PD, including protein aggregation, mitochondrial dysfunction, and disrupted autophagy-lysosomal mechanisms. It is likely that the contribution of these pathogenic processes varies between individuals and this may account for some of the clinical heterogeneity observed in the symptoms and rate of progression of PD. Although rarely considered in clinical trial design, both clinical and biological heterogeneity of study cohorts may have a major impact on outcomes, particularly in small scale trials. Lack of phenotypic stratification may have contributed to negative outcomes in some of the previously completed early-phase trials in this field. In future trials of immunosuppressive and anti-inflammatory therapies in PD, selecting participants based on inflammatory phenotypes using baseline biomarker analyses could potentially enhance success.
APPENDIX: SUMMARY OF IMMUNOSUPPRESSIVE/ANTI-INFLAMMATORY AGENTS UNDER ACTIVE INVESTIGATION
The eight clinical trials of immunosuppressive/anti-inflammatory drugs which are either active or recently completed are listed in Table 1 and summarized in more detail in the section below.
Background: Azathioprine is a peripherally acting immunosuppressant with a broad action on immune cell populations, which is widely used in clinical practice, with substantial evidence of efficacy in a range of inflammatory and autoimmune conditions, including diseases of the central nervous system.99,100, 99,100 It acts on purine synthesis, leading to a reduction in lymphocyte counts in peripheral blood. 101 As discussed in this review, there is abundant evidence that the peripheral immune system is activated in PD with alterations in multiple lymphocyte subsets, and that lymphocytes infiltrate into the central nervous system in PD.
A clinical trial repurposing azathioprine as a potential disease modifying therapy for PD was commenced in 2021 and completed in February 2024 but results are not yet available. This proof of concept study explores the hypothesis that peripheral immune activation is a significant driver of central immune activation and disease progression through specifically targeting the peripheral immune component.
Title: AZAthioprine immunosuppression and disease modification in Parkinson’s Disease (AZA-PD): a randomised double-blind placebo-controlled phase II trial
Phase: 2
Status: Completed, analysis in process
Clinical Trial ID: ISRCTN14616801
Sponsor: Cambridge University Hospitals NHS Foundation Trust and University of Cambridge
Study Design: This is a randomized double-blind placebo-controlled trial, at a single site. 67 participants with early PD (<3 years duration) and no immune or inflammatory comorbidities have been recruited and were randomized 1 : 1 to azathioprine 1–2 mg/kg or placebo administered orally for a treatment period of 12 months, with a further 6 months follow up period post treatment cessation.
Outcome measures: The primary outcome measure is change in the gait/axial subscore of the MDS-UPDRS over the 12-month treatment period. This is a measure which is proposed to be the most sensitive component of the MDS-UPDRS to disease progression and relatively resistant to changes in dopaminergic therapy. 102 Other exploratory clinical outcomes include the change in total MDS-UPDRS, Addenbrookes cognitive examination III (ACEIII), Geriatric depression scale (GDS), Parkinson’s Disease Quality of Life Questionnaire-39 (PDQ-39) and Non-motor symptom scale (NMSS). In addition, the trial includes “proof of mechanism” exploratory outcomes investigating the treatment effect of azathioprine on peripheral and central measures of immune activation. These include immunophenotyping of peripheral blood and cerebrospinal fluid, inflammatory cytokine analysis and [11C]-PK11195 PET/MR brain imaging before and after treatment.
Comments: The results of this study are expected within the next few months. Whilst it is a relatively small trial, it has a robust double-blind placebo-controlled design, with a carefully selected primary endpoint to maximize the chances of detecting a change in disease progression. The inclusion of detailed biofluid and imaging measures will provide insight into the interaction between peripheral and central immune compartments in PD. 77
Background: Sargramostim is a human recombinant granulocyte-macrophage colony-stimulating factor (GMCSF) that can increase T regulatory immune cell numbers and function. It is currently FDA approved for myeloid reconstitution following chemotherapy or bone marrow transplants.
The adaptive immune response has been strongly implicated in PD, with changes in both pro-inflammatory and regulatory lymphocyte subsets. In particular, a reduction in T regulatory cells has been reported with an impaired ability to suppress the proliferation of T effectors cells.27,31,103, 27,31,103 Increasing T regulatory cells through treatment with sargramostim mitigates inflammation, microgliosis and neuronal death in MPTP mice models of disease.104,105, 104,105
In 2016, a phase 1 randomized double blind, placebo-controlled trial of sargramostim (NCT01882010) was completed. Twenty PD patients were randomized in a 1:1 ratio to receive sargramostim at a dose of 6 μg/kg/day administered subcutaneously or placebo for 56 days. 78 The primary outcomes were safety and tolerability, and the trial was not powered to assess clinical efficacy. Adverse events were reported in all participants in the treatment arm, with the most common being injection site reactions, abnormal blood results and bone pain. 78 There was evidence of the anticipated effect on immune populations, with an increase in numbers of T regulatory cells and functional assays showing a restoration in their ability to suppress T effector cells. 78
Phase 1 testing of sargramostim has recently resumed but at a lower dose due to concerns regarding the tolerability of the drug at the initial doses used. Two related trials from the same academic team are currently underway.
Titles: Safety, Tolerability and Biomarker Assessments of Leukine (Sargramostim) During Extended Timed Treatment for Parkinson’s Disease: A Phase I Pilot Study (Trial 1)
A Phase 1, Open-label Study to Validate Treat-ment-induced Biomarkers Following Sargramostim Treatment in Parkinson’s Disease (Trial 2)
Phase (both): 1
Status (both): Active – not recruiting
Clinical Trial ID’s: NCT03790670 (Trial 1); NCT05677633 (Trial 2)
Sponsor (both): University of Nebraska
Study Design: Trial 1 is an open label single center study evaluating 3 μg/kg/day of sargramostim in 5 participants with PD, treated for a period of 36 months. Trial 2 is a further 48-week open label study of 3 μg/kg/day sargramostim in 11 participants with PD aiming to further validate biomarker and safety outcomes, and projected to complete in 2024.
Outcome measures: The primary outcome measures for both clinical trials are the safety and tolerability of 3 μg/kg/day of sargramostim, administered for 5 days followed by a 2-day drug holiday for a prolonged treatment duration of either 36 months or 48 weeks, assessed using clinical and biochemical measures. Secondary outcome measures will assess the effect of treatment on immune cells subsets and function before, during and on cessation of treatment. Clinical efficacy will also be assessed as a secondary outcome, monitoring change in MDS-UPDRS parts 1–4scores.
Results: Initial results from Trial 1 with analyses at a 1-year time point and a 33-month time point with a 3-month trial drug ‘break’ at 24 months have been published.79,80, 79,80 With the lower doses used, and the 2-day drug free holiday per week, there was a reduction in the frequency and severity of severe drug related adverse events.79,80, 79,80 The MDS-UPDRS part III improved on treatment, with a return to baseline when the trial drug was stopped for a period of three months.79,80, 79,80 T regulatory cell numbers showed a sustained increase during treatment and although anti-drug antibodies developed, they were not thought to impact on treatment effectiveness.79,80, 79,80
Comments: The clear limitations of these trials are their small sample size and lack of placebo arm, which need to be considered when interpreting the reported clinical outcomes. In addition, a heterogenous PD cohort has been recruited in Trial 1 with a range in disease duration from 3–14 years. Although the trial has shown that sargramostim is effective at boosting T regulatory cell numbers across disease stages, there is evidence to support the hypothesis that immune activation is most prominent early in the disease course29,106, 29,106 and therefore an early-stage cohort with power to explore treatment effect on disease progression will be important. With the need to self-administer subcutaneous injections 5 times weekly assessing tolerability and optimum dosing in the PD population for extended treatment durations is needed prior to larger clinical trials. The development of a novel formulation of GMCSF, PDM608, which has a longer treatment duration, is intended to mitigate some of the injection burden. 81
Background: Montelukast is cysteinyl leukotriene receptor type 1 (CysLT1) antagonist and is currently licensed for use as an adjuvant therapy in asthma. Leukotrienes play an important role in the inflammatory response, mediating lymphocyte migration, vascular permeability and astrocyte proliferation 107 when bound to CysLT1 .
In models of CNS disease, including MPTP induced mouse models of PD, CysLT1 expression is increased. 108 Inhibition of this pathway with montelukast can attenuate dopaminergic neuronal loss, reduce microglial activation and reduce the production of pro-inflammatory cytokines TNFα and IL1β in multiple animal models of PD.82,83,109, 82,83,109 Inhibition and knockout of 5-lipo-oxygenase, which is essential in the production of leukotrienes is also neuro-protective.110,111, 110,111
To investigate the potential of montelukast as a disease modifying therapy in PD, a small trial assessing safety and tolerability was completed in 2022.
Title: Effects of Montelukast on neuroinflammation in Parkinson’s disease. An open-label single-center trial.
Phase: 2
Status: Completed
Clinical Trial ID: EUCTR2020-000148-76-SE
Sponsor: Stockholm Health Care Services
Study Design: This was a small, open label single arm trial in which 15 participants with PD with a Hoehn and Yahr of≤2 were treated with montelukast oral tablets 20 mg twice daily for a period of 12 weeks at a single center in Sweden. Participants continued their normal levodopa medications.
Outcome measures: The primary outcome measure was the safety and tolerability of montelukast in patients with PD assessed through numbers of adverse events noted at five time points throughout the trial. Secondary outcome measures included inflammatory biomarkers in the plasma and CSF, changes in [11C]PBR28 TSPO PET brain imaging to assess microglial activation, levels of montelukast in the CSF, urine, and blood at 4 weeks and changes in MDS-UPDRS, NMSS, PDQ-39 and Montreal Cognitive Assessment (MoCA) scores between baseline, 4 weeks of treatment and 12 weeks oftreatment.
Results: Results have been made available through the EU clinical trials register. All participants completed the trial with 1 serious adverse event which was not treatment related and 16 non-serious adverse events. 6 of these were attributed to trial specific investigations such as lumbar punctures and PET imaging. Loose stools were the commonest treatment-related adverse event with 20% of participants affected. Grey matter binding of [11C]PBR28 TSPO PET imaging showed no statistically significant change at the end of treatment in comparison to baseline. Changes in MoCA and MDS UPDRS part I–III scores showed a significant improvement following 12 weeks of montelukast treatment. No significant changes were noted in the NMS, PDQ-39 and part 4 of the MDS-UPDRS. Blood and CSF biomarker results are not yet available.
Comments: The improvement in clinical measures (MDS-UPDRS and MoCA) is difficult to interpret given the lack of a placebo group in this study, and the likelihood of practice effects on the MoCA. Although no effect on brain inflammation was demonstrated, primary endpoints of safety were achieved and a larger randomized placebo-controlled trial has been approved using a novel montelukast formulation that can be administered as a thin biofilm orally, negating the need for swallowing multiple tablets daily, which will be useful in PD patients with dysphagia. 84
Background: NE3107 is an orally active drug that penetrates the blood-brain barrier. 112 It has anti-inflammatory actions through the binding and regulation of extracellular signal-regulated kinase (ERK) mediated inflammatory pathways, leading to a reduction in the transcription of pro-inflammatory genes such as IL-1β, IL-6 and TNF-α. 85
ERK is a part of the mitogen activated protein kinase family that has an essential role in coordinating cellular responses to external stimuli. 113 In human PD and DLB brains, cytoplasmic inclusions of phosphorylated ERK have been found in neuronal cells of the substantia nigra, even in early disease stages. 114 ERK mediated pathways have also been implicated in mouse models of levodopa induced dyskinesias, further supporting targeting this pathway in PD. 115
In an MPTP mouse model of PD, NE3107 attenuated dopaminergic neuronal loss, preserved motor abilities on the rotarod test, and reduced expression of proinflammatory mediators such as iNOS, TNF-α, and IL-1β. 112 In a marmoset model of PD, NE3107, administered after MPTP, was neuroprotective and led to improvements in mobility in comparison to placebo. 116 This effect was increased with the co-administration of levodopa and a reduction in levodopa induced dyskinesias was observed. 116
A phase 2 trial investigating NE3107 in PD completed in January 2022.
Title: A Double-Blind Placebo Controlled Safety Tolerability and Pharmacokinetic study in Parkinson’s Disease Participants Treated with Carbidopa/Levodopa and NE3107
Phase: 2
Status: Completed, analysis in process
Clinical Trial ID: NCT05083260
Sponsor: BioVie Inc.
Study Design: This was a multicenter double-blind placebo-controlled study in patients with Parkinson’s disease assigned in a 1 : 1 ratio to placebo or 20 mg NE3107 twice daily for a period of 28 days. The target sample size was 40 participants, with actual recruitment of 46 participants, on at least 300 mg of levodopa/carbidopa with a history of motor fluctuations and early-morning off periods.
Outcome measures: The primary outcome was safety and tolerability of oral NE3107 as an adjuvant therapy in patients with PD on treatment with levodopa. Maximum plasma concentrations of NE3107, and levodopa, administered alone or in combination were also measured. Efficacy measures included changes in MDS-UPDRS scores in the ON and OFF state, length of time in the defined ON and OFF states determined by patient self-categorization diaries, time from administration of levodopa to the ON state, and non-motor symptoms and dyskinesia assessments.
Results: Initial results were presented at the 2023 International Conference of Parkinson’s Disease and Movement Disorders. 86 NE3107 was reported to be well tolerated with no drug related adverse events observed. At the end of the 28-day treatment period, an improvement of > 30% in MDS-UPDRS-III measured two hours post levodopa was noted in 80% of the treatment arm (receiving NE3107 + levodopa) compared to 63.6% of the placebo arm (receiving placebo+levodopa), but the statistical significance of this finding was not reported. NE3107 did not alter the pharmacokinetic profile of levodopa but a significant number of participants treated with NE3107 had a reduction in early morning OFF periods.
Comments: The preliminary results suggest that NE3107 is safe and well tolerated, and may improve motor function independent of levodopa concentrations, implying that NE3107 might warrant investigation as an adjunctive symptomatic therapy in PD. This trial was not designed to investigate disease modification, given the short duration of treatment. Publication of the full results is still awaited, and assessing target engagement of NE3107 with inflammatory biomarkers will be important to help guide whether progression to phase 3 trials is warranted
Background: AKST4290 is a potent small molecule antagonist of CCR3, a chemokine receptor for eotaxin (CCL11), which is expressed on T cells, amongst other leukocytes (basophils, monocytes and eosinophils) and involved in T cell migration into the brain. 88 CCL11 and CCR3 have been shown to increase with aging and increased levels are associated with cognitive decline.117,118, 117,118 AKST4290 does not penetrate the blood brain barrier to a pharmacologically significant level, but it is assumed to act on peripheral T cells. 88 AKST4290 reduces central T cell infiltration and microgliosis in mice immunized with myelin oligodendrocyte glycoprotein (which induces marked T cell infiltration into the brain), and has been shown to reduce the cognitive and motor deficits seen in naturally aged mice; 88 as such it is a target of interest for neuroinflammatory diseases including PD.
The evidence supporting the role of CCR3/CCL11 in PD pathogenesis is variable. Serum and CSF levels of CCL11 and expression of CCR3 on peripheral eosinophils are not increased in PD in comparison to controls,119–121 but levels of CCL11 are increased in microglial cells of the substantia nigra in post-mortem PD human and MPTP mice brains. 122 Treating MPTP mice with peripheral neutralizing antibodies to eotaxin and RANTES/CCL5 (another chemokine which also binds to CCR3 with medium affinity) reduced inflammatory cytokine production, peripheral T cell infiltration into the substantia nigra and dopaminergic neuronal death leading to an improvement in motor deficits, 122 further supporting targeting CCR3 in PD.
A clinical trial assessing the efficacy and safety of AKST4290 was completed in 2021.
Title: Study Assessing Efficacy and Safety of AKST4290 in Subjects with Parkinson’s Disease on Stable Dopaminergic Treatment (TEAL)
Phase: 2
Status: Completed, analysis in process
Clinical Trial ID: NCT04369430
Sponsor: Alkahest Inc
Study Design: This was a randomized multicenter double-blind placebo-controlled trial evaluating the efficacy and safety of AKST4290 400 mg twice daily compared to placebo (1 : 1) in 110 participants with PD. Treatment duration was 12 weeks.
Outcome measures: The primary outcome measure was change in MDS-UPDRS part 3 motor scores measured in the ‘OFF’ phase between baseline and week 12. Secondary outcome measures were safety of the trial drug (assessed via review of adverse events, changes in vital signs, clinical laboratory parameters and ECGs) and further clinical efficacy outcomes as measured by change in the following measures over the treatment period: total MDS-UPDRS in the ‘ON’ state, MOCA, Schwab and England Activities of Daily Living Scale, Clinical Impression of Severity Index-PD (CISI-PD), PDQ-39, Sheehan Suicidality Tracking scale, a 10 m timed walk score and mean time spent with and without dyskinesia using the Hauser 3-Day Patient Diary.
Results: 110 participants were recruited through multiple sites. All participants had a Hoehn and Yahr stage of≤2.5 and were on stable dopaminergic therapy, with motor symptoms worsening in the ‘OFF’ state. Participants had a mean PD disease duration at baseline of 5.7 years in the treatment group and 7.6 years in the placebo group. The results of the study have been posted on ClinicalTrials.gov (NCT04369430). Similar reductions in the motor subscore of the MDS-UPDRS were seen in participants on AKST4290 and placebo, in both the ON and OFF state. There were no improvements in PDQ-39 and MDS-UPDRS part II scores (motor experiences of daily living) in the active treatment group. Two patients in the treatment arm and three in the placebo arm withdrew due to adverse events and there was one case of sudden cardiac death in the treatment arm. It is unclear whether this was treatment related. Publication of the final results is awaited.
Comments: Given the short treatment duration, it is perhaps not unexpected that this trial did not meet its primary endpoint of clinical efficacy. Clinical heterogeneity within the cohort may also have contributed to this negative result.
Background: Inflammasomes are intracellular multimeric protein complexes which amplify the inflammatory response in innate immune cells. They are characterized by their pathogen recognition receptor molecule. The NLRP3 inflammasome has been highly implicated in PD and has been shown to be activated by pathogenic forms of alpha-synuclein.14,15, 14,15 Activation leads to downstream release of inflammatory cytokines including IL-1β and IL-18, leading to the propagation of a pro-inflammatory environment. 16 NLRP3 knockout is protective against MPTP induced dopaminergic neuronal loss, microglial activation and midbrain cytokine production, 123 and administration of NLRP3 inhibitors in alpha synuclein models of disease attenuates dopaminergic neuronal loss. 124 Levels of NLRP3 and its downstream cytokine IL-1β in the plasma positively correlate with motor symptom severity in PD, 125 and increases in NLRP3 protein expression in postmortem tissue have been reported not only in PD 13 but also in cases with asymptomatic Lewy pathology. 126 This suggests that NLRP3 inflammasome activation might predate symptom onset and increase with disease progression, making it a strong candidate for therapeutic targeting.
Background: Selnoflast (R07486967) is an orally administered selective NLRP3 inflammasome inhibitor and has been studied in a phase 1 trial in healthy controls (NCT04086602, no published results) and ulcerative colitis with a favorable pharmacokinetic and safety profile at an oral dose of 450 mg daily. 127
Although no positive pharmacodynamic results were recorded in vivo in clinical human trials published to date, immune cells from patients receiving selnoflast which were then stimulated with LPS showed a reduction in IL-1β release, suggestive of successful NLRP3 inflammasome inhibition. 127 Unpublished data, which is referenced in the ulcerative colitis clinical trial publication 127 indicates that selnoflast has a selective inhibitory effect on only the NLRP3 inflammasome, with no effects on the Nucleotide-binding Oligomerization Domain (NOD)-like Receptor (NLR) family CARD domain containing 4 (NLRC4) or Absen t In Melanoma-2 (AIM2) inflammasome. Selnoflast is now being trialed in a phase 1 study in PD with anticipated completion in 2025.
Title: A Phase 1b, Adaptive, Multi-Center, Randomized, Double Blind, Placebo-Controlled, Parallel Design Study to Investigate The Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of RO7486967 in Participants With Early Idiopathic Parkinson’s Disease
Phase: 1
Status: Active – recruiting
Clinical Trial ID: NCT05924243
Sponsor: Roche
Study Design: This is a multi-center randomized double-blind placebo-controlled trial. Seventy-two participants will be randomized on a 2 : 1 basis to receive selnoflast 200 mg twice daily or placebo for a period of 28 days.
Outcome measures: The primary outcomes are safety and tolerability, assessed through the percentage of participants with adverse events, abnormal laboratory blood and urine findings, vital signs, ECG parameters and suicidal ideation using the Columbia Suicide Severity Rating Scale. Secondary outcome measures will investigate the pharmacokinetics of selnoflast in plasma and its anti-inflammatory actions in the brain, assessed using [18F]-DPA-714 TSPO PET imaging pre and post treatment.
Comments: Results of this phase 1 trial are expected in 2025. It is not established whether this drug crosses the blood brain barrier in humans and CSF selnoflast levels are not being evaluated in this study. However, TSPO-PET brain imaging is being used as a measure of target engagement and will provide important data on the impact of this inflammasome inhibitor on neuroinflammation. The short trial duration will facilitate rapid evaluation of primary safety outcomes, and if positive, will inform a longer duration trial to assess clinical efficacy.
Background: NT-0796 is a novel orally available selective NLRP3 inflammasome inhibitor with good CNS penetration. 128 Preclinical studies have shown a low toxicity profile in rodents, 128 and in vitro studies show NT-0796 reduces IL1β release from stimulated human PBMCs. 128 In a mouse model of obesity, NT-0796 reduced hypothalamic astrogliosis. 129 There are no published data on the effects of NT-0796 in pre-clinical models of PD. Nodthera announced in 2021 that the first healthy volunteers were trialed on oral NT-0796, and recently issued a press release reporting positive results in a phase 1b/2a study in PD patients. 90 Interim results have been presented at the AD/PD conference in March 2024.
Title: Unavailable
Phase: 1b/2a
Status: Completed, analysis in process
Clinical Trial ID: Unavailable
Sponsor: Nodthera
Study Design: This is an open label study involving two cohorts of participants. The first cohort of four healthy volunteers received oral NT-0796 twice daily for seven days, and the second cohort of up to 10 patients with PD received the same dose for 28 days.
Outcome Measures: The primary outcome measures were safety and tolerability of NT-0796 in healthy volunteers and patients with PD. Secondary objectives included assessing the pharmacokinetic and pharmacodynamic profile of the agent using blood and CSF biomarkers. 130
Results: Results have been presented at the 2024 AD/PD conference 130 and in a recent press release. 90 Adverse events were reported to be mainly mild and transient, with no serious adverse events observed. In healthy volunteers, there was a reduction in plasma levels of IL1β, IL18, and CXCL1 within 7 days. A reduction in CRP in those participants with elevated levels at baseline was also observed in both healthy volunteers and PD participants by day 7. 130 CSF analysis confirmed blood-brain barrier penetration with maintenance of NT-0796 levels for over 24 hours, and a reduction in both inflammatory cytokines and levels of neurofilament light chain and soluble TREM2 in PD participants over 28 days treatment.
Comments: While the full results are not yet available, the reported positive effects of NT-0796 on inflammatory and neurodegeneration biomarkers are encouraging. Further safety and tolerability data is needed in larger numbers of participants to support advancement of this compound and phase 2 studies are being planned.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
BP is supported by the National Institute of Health Research, UK (NIHR). JCG was supported by the Cambridge Centre for Parkinson-Plus. CHWG is supported by the Medical Research Council, UK (MR/W029235/1) and the NIHR Cambridge Biomedical Research Centre (NIHR203312). The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care. The authors also receive grant support from Cure Parkinson’s, Parkinson’s UK, the MRC, the Rosetrees Trust, and Addenbrooke’s Charitable Trust.
CONFLICT OF INTEREST
The authors have no conflicts of interest to report.