
Abstract
Dysregulation of innate and adaptive immunity can lead to alpha-synuclein (α-syn) misfolding, aggregation, and post-translational modifications in Parkinson’s disease (PD). This process is driven by neuroinflammation and oxidative stress, which can contribute to the release of neurotoxic oligomers that facilitate dopaminergic neurodegeneration. Strategies that promote vaccines and antibodies target the clearance of misfolded, modified α-syn, while gene therapy approaches propose to deliver intracellular single chain nanobodies to mitigate α-syn misfolding, or to deliver neurotrophic factors that support neuronal viability in an otherwise neurotoxic environment. Additionally, transformative immune responses provide potential targets for PD therapeutics. Anti-inflammatory drugs represent one strategy that principally affects innate immunity. Considerable research efforts have focused on transforming the balance of pro-inflammatory effector T cells (Teffs) to favor regulatory T cell (Treg) activity, which aims to attenuate neuroinflammation and support reparative and neurotrophic homeostasis. This approach serves to control innate microglial neurotoxic activities and may facilitate clearance of α-syn aggregates accordingly. More recently, changes in the intestinal microbiome have been shown to alter the gut-immune-brain axis leading to suppressed leakage of bacterial products that can promote peripheral inflammation and α-syn misfolding. Together, each of the approaches serves to interdict chronic inflammation associated with disordered immunity and neurodegeneration. Herein, we examine research strategies aimed at improving clinical outcomes in PD.
Keywords
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disorder after Alzheimer’s disease. PD prevalence is 1–3% of those 65 years of age or older and is projected to affect over 1.2 million people by 2030 [1–3]. While the etiology and driving forces that govern progression of PD remain elusive, factors such as genetics, environment, and inflammatory processes serve as disease risk factors linked to degeneration of the dopaminergic neurons that originate in the substantia nigra (SN) pars compacta and innervate to the striatum [4–8]. Moreover, dopaminergic neuronal loss and consequent depletion of the neurotransmitter, dopamine are associated with PD signs and symptoms. These include tremor, slowed movement (bradykinesia and hypokinesia), rigidity, mask-like facial expressions (hypomimia), rigidity of limbs, neck, and trunk, and impaired posture and balance [9, 10]. These are insidious and associated with progressive neuronal loss that precede clinical disease signs and symptoms [11].
As the prototypical synucleinopathy, a pivotal disease hallmark in PD is the cytoplasmic accumulation of proteinaceous aggregates or Lewy bodies (LBs) comprised primarily of misfolded α-synuclein (α-syn) and ubiquitin [12]. The contribution of LBs to disease is substantive, including disruption of pre-synaptic communications, oxidative stress, mitochondrial toxicity, and intraneuronal proteomic homeostasis [13]. The release of α-syn from neurons and interaction with microglia, activate those microglia to inflammatory states. Neuroinflammation and oxidative stress fuels α-syn modification, misfolding, and oligomerization, which promote LB generation. α-Syn-mediated microglial activation results in production and secretion of pro-inflammatory cytokines and mediators of neurotoxicity, thus these reactive microglia become proximal sources for protein modification and dopaminergic neuronal injury and cause even greater release of pathogenic forms of α-syn [14–16]. Such pathogenic events over time perpetuate α-syn misfolding, aggregation, and eventual neurodegeneration [17]. However, events associated with this progressive cycle may prove amenable to therapeutic interventions designed to slow disease [13, 18–21].
CURRENT THERAPEUTICS
Clinical treatments are currently limited in large part to dopamine replacement strategies that include use of the dopamine prodrug levodopa (L-DOPA) and dopamine receptor agonists [22]. Both serve to either replenish or mimic dopamine at the synaptic junction to enhance post-synaptic dopamine receptor activation. These approaches serve to affect the dopaminergic circuits responsible for movement coordination [23–25]. Alternatively, stimulation of compromised neuronal networks can be accomplished through surgically implanted probes by deep brain stimulation (DBS) [17, 22]. In essence, current clinical approaches to treatment of PD provide symptomatic relief, thus are not specifically designed to target modifications of pathological disease progression [23, 26].
DISEASE-MODIFYING THERAPIES (DMTs)
The recent focus on DMTs in PD remains an intense area of research given the projected increase in PD prevalence [27, 28]. One DMT targets α-syn misfolding and aggregation linked to the inflammatory release of neurotoxic factors by microglia, which induce oxidative stress on neighboring neurons and further compromise the neuroprotective profile of the brain through aggregated α-syn accumulation and spread (Fig. 1). Additionally, innate and adaptive immunity that lead to neuroinflammation are established contributors to PD neurodegeneration. Therefore, therapies that target innate microglial inflammation in PD using nonsteroidal anti-inflammatory drugs (NSAIDs), anti-inflammatory cytokines, and/or agents that reduce inflammasome activation have been explored (Fig. 1) [15, 29–32]. Declining regulatory control allows perpetuation of low-level neurotoxic pro-inflammatory cytokines and mediators of oxidative stress. Contrastingly, control of effector T cells (Teffs) and reactive microglia through induction or amplification of Treg numbers and function, or by engineering chimeric antigen receptor (CAR)-Tregs, have also gained interest. In all, mechanisms that correct the diminishing regulatory adaptive immunity, which fails to maintain a healthy tolerogenic environment in the central nervous system (CNS), may prove most beneficial and necessary for neuronal replacement therapy. Indeed, evidence of LB pathology and proximate inflammatory responses observed in tissues from trials of neuronal transplants underscore the importance of immune responses in non-autochthanous cell processes of dopaminergic neurodegeneration [33, 34]. Oxidative stress within the brain microenvironment can lead to post-translationally modified and misfolded α-syn that damages mitochondria, activates microglia and ultimately, diminishes dopaminergic neuronal vitality and the integrity of the striatal innervations [16, 36]. Under normal conditions, microglia serve to clear misfolded protein aggregates that emerge as a result of disease or injurious event(s), but under disease conditions, they become chronically activated within the pro-inflammatory environment and provide a driving force for the perpetuation of protein misfolding and neuronal damage [37]. Thus, primary targets for DMT strategies could include not only reactive microglia, but also the overproduction of pro-inflammatory factors such as tumor necrosis factor alpha (TNF-α), interleukin-1 beta (IL-1β), IL-6, nitric oxide, prostaglandin E2, reactive oxygen species, and the enzyme nicotinamide adenine dinucleotide phosphate oxidase (NOX) [38–41]. In conjunction with targeting inflammation, suppression of oxidative modification and its processes would benefit the intervention of α-syn misfolding, aggregation, and spreading (Fig. 1).
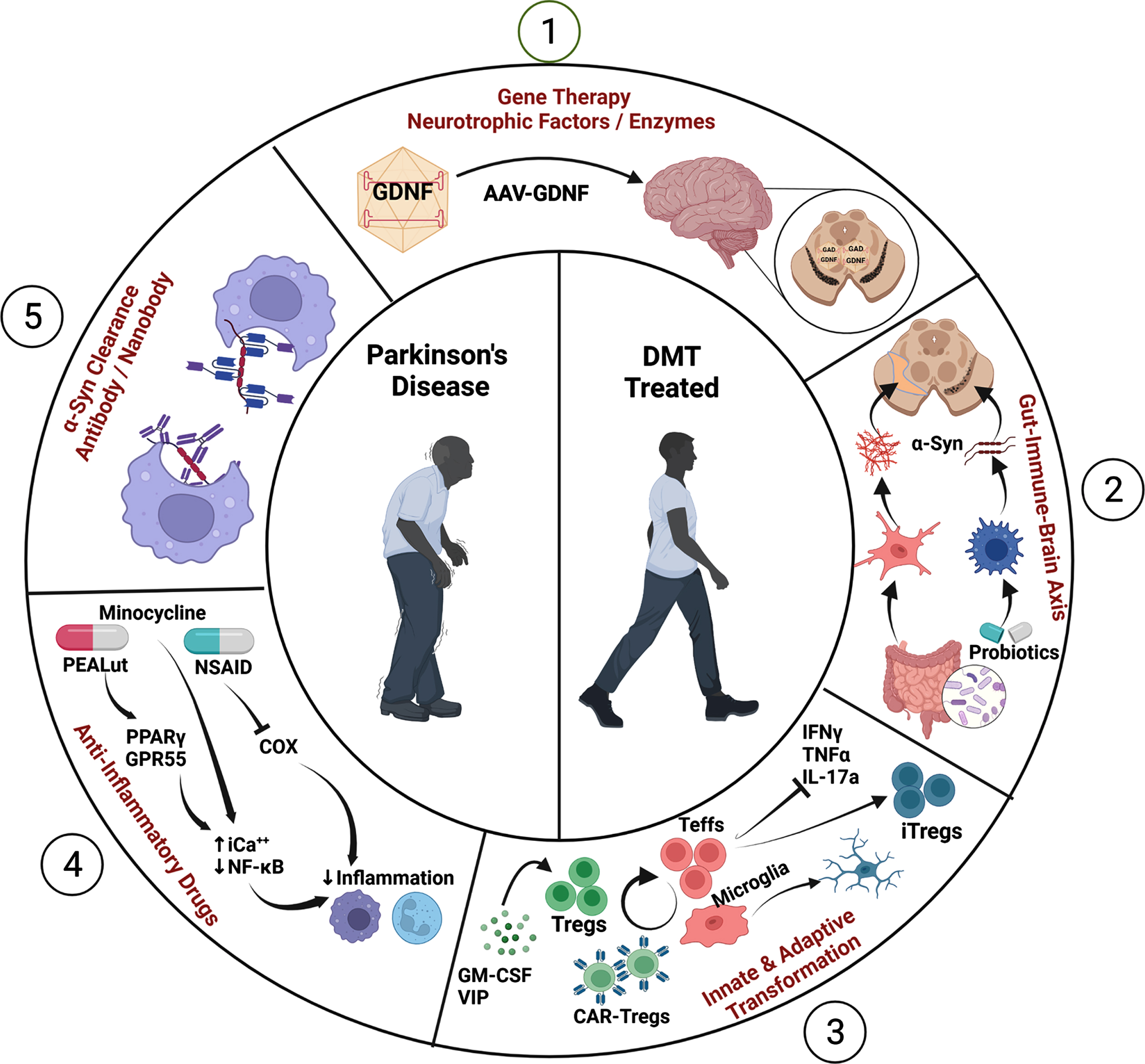
Mechanisms of Disease-Modifying Therapies (DMTs). (1) Gene therapy delivers neurotrophic factors and enzymes in disease-affected brain to improve outcomes as demonstrated in pre-clinical PD studies. (2) Probiotics transform intestinal and peripheral immune profiles, which control α-syn folding and brain microenvironments. (3) CAR-Tregs or Tregs elicited from immune modulators suppress reactive M1 microglia and Teffs to inhibit proinflammatory cytokine production and promote transformation of M2 microglia and induced Tregs (iTregs) which yield a neuroprotective environment. (4) Anti-inflammatory drugs such as NSAIDs, and PEALut, and minocycline inhibit pro-inflammatory activities in the periphery and brain. (5) Vaccines, antibodies, and nanobodies directed at α-syn polymers serve to clear or prevent aggregates of neurotoxic α-syn fibrils and oligomers and protect dopaminergic neurons. Created with BioRender.
DMT IN INNATE IMMUNITY
Evidence of reactive microglia in post-mortem brains of PD patients and in preclinical models have been appreciated for several decades and suggest a prominent role of chronic inflammation in PD [41–46]. Bioimaging using PET radioligands, such as PK11195 to detect the upregulated 18kDa translocator protein (TSPO) by activated microglia [47], showed increased binding in the midbrains of PD patients compared to controls [48–50]. Quantitation of PK11195 PET scans were negatively correlated with the loss of neurons expressing the dopamine transporter and positively correlated with motor severity as determined by the Unified Parkinson’s Disease Rating Scale (UPDRS) scores for movement [48]. Consistent detection of increased pro-inflammatory cytokine levels from serum, cerebrospinal fluid, and tissues of patients also underscore the chronic nature of systemic inflammation in PD. Abundant evidence indicates that high levels of chronic inflammation lead to increased oxidative states, and play a major role in disease progression and possibly etiology [51, 52]. Activated microglia secrete neurotoxic mediators and sufficiently increase oxidative stress to induce protein modifications and misfolding of α-syn, which is then secreted or released into the extraneuronal environment upon cell injury or death and activates surrounding microglia to perpetuate a chronic inflammatory state. Indeed over-activation of microglia significantly upregulates expression of a large group of pro-inflammatory cytokines, including TNF-α, interleukin-6 (IL-6), IL-1β, and interferon gamma (IFN-γ), which contribute to the acceleration of nigral dopaminergic neuronal degeneration. Moreover, inflammatory and oxidative mediators produced by activated microglia upregulate the expression of monomeric α-syn in neurons and promote its aggregation and spread of pathogenic forms of α-syn to other neurons [53]. In addition, immune forces that control and regulate microglia and chronic inflammation are diminished in PD [52, 54–56].
With an abundance of evidence supporting inflammatory processes in PD, applying anti-inflammatory DMTs in PD seems warranted. NSAIDs are targeted to suppress infiltrating peripheral immune responses and alleviate tissue damage associated with hyperactive immunity (Fig. 1). Findings in a longitudinal study designed to assess the likelihood of developing PD and NSAID usage showed significant reductions in disease prevalence when the drugs were taken at least twice a week [57–59]. However, not all NSAIDs afforded decreased risk and were limited to specific compounds and dosing regimens. Notably, ibuprofen has been independently verified as a potential disease-modifying medicine [60, 61]. A recent study showed that aspirin and ibuprofen may lower the risk of leucine-rich repeat kinase 2 (LRRK2)-linked PD and provide a simple DMT for people exhibiting the LRRK2 mutation [62]. Interestingly, some NSAIDs such as ibuprofen and indomethacin have been reported to display PPAR-γ agonist activities as well (vide infra) [63–65]. In contrast, no association between the usage of NSAIDs and decreased risk of developing PD could be determined in a meta-analysis of 15 studies of NSAIDs use in PD [66]. A similar finding reported that non-aspirin NSAIDs had no association to PD in a population-based control study, which included a cohort of 206 newly diagnosed idiopathic PD subjects [67]. Nonetheless, due to existing discrepancies in the findings, a relationship between NSAIDs use and risk of PD remains controversial.
Another strategy advances the use of minocycline to attenuate microglial responses [68]. Minocycline is a second-generation, high-lipophilic tetracycline that easily crosses the blood-brain barrier (BBB), and thus, can accumulate within the CNS at levels 5-fold higher than other tetracyclines [69]. It is a potent inhibitor of microglial activities and immune cell activation, which results in the reduced production of proinflammatory cytokines, such as TNF-α, IL-1β, and IL-6 [70]. Microglial signaling pathways affected by minocycline include inhibition of p38 mitogen-activated protein kinase (MAPK) and polarization to M1 phenotypes by STAT1/STAT6 pathways [69]. Many pre-clinical studies have shown that minocycline attenuates microglia or microglial activities, reduces neuroinflammation and oxidative stress, and increases neuroprotection [71–80]. Contrastingly, in two studies, minocycline treatment of MPTP-intoxicated mice and cynomolgus monkeys increased dopaminergic neurodegeneration, yet still reduced reactive microglia [75, 81]. With an abundance of evidence supporting its therapeutic potential, a phase II clinical trial tested minocycline and creatine in early PD, and found that administration of minocycline or creatine for 18 mounths failed to slow disease progression compared to placebo controls [82, 83].
Another therapeutic target of considerable interest is the family of peroxisome proliferator-activated receptors (PPARs). PPARs are ligand-inducible transcription factors belonging to the hormone nuclear receptor superfamily in which three PPAR isoforms have been identified as PPAR-α, PPAR-β/δ, and PPAR-γ [84]. PPAR ligation leads to heterodimerization with retinoid X receptors (RXRs), and subsequent recognition and binding PPAR response elements (PPRE) within promoter regions [85]. This leads to transcriptional regulation of a variety of processes including cell proliferation and differentiation, fat and carbohydrate metabolism, inflammation, vascular biology, and cancer [86]. Of the three isoforms, PPAR-γ appears most relevant to inflammation and neurodegeneration as it is expressed not only in adipose tissue, but also by immune cells, such as myeloid cells, dendritic cells, and T cells, including Tregs [87, 88]. Of interest, PPAR-γ also co-localizes with tyrosine hydroxylase expression in dopaminergic neurons [89], and PPAR-γ agonists have been shown to attenuate dopaminergic signaling [90]. More importantly, some synthetic PPAR-γ agonists, known as thiazolidinediones (TZD) or glitazones that are used to control hyperglycemia in type-2 diabetes, also exhibit anti-inflammatory properties [91, 92]. Pioglitazone, a potent TZD PPAR-γ activator, reduces microglia activation and pro-inflammatory cytokine production [93, 94]. This mechanism has been attributed to exertion of PPAR-γ-mediated inhibition of pro-inflammatory transcription factors such as NF-κB, AP-1 and STAT1 [95, 96]. Moreover, PPAR-γ agonists have been shown to be neuroprotective in models of PD wherein reactive microglia concomitantly show diminished activation [97–102]. Interestingly, PPAR-γ agonists can polarize activated microglia from a M1 phenotype to a M2 phenotype with diminution of proinflammatory cytokine expression and upregulation of anti-inflammatory cytokines [93, 103]. However, in a phase II clinical trial in early PD, treatment with either 15 mg/day or 45 mg/day of pioglitazone or placebo failed to meet primary outcome to change the total UPDRS score from baseline, thus suggesting that at the dosages studied, pioglitazone was unlikely to modify disease progression in PD [104].
A recent anti-inflammatory therapeutic candidate is co-ultramicronized PEALut (co-ultra PEALut) which consists of two compounds, palmitoylethanolamide (PEA) and the flavonoid, luteolin (LUT) [105] (Fig. 1). Both are anti-inflammatory and neuroprotective agents, which exhibit synergistic activities when combined [106], while PEA has been shown to serve as a PPAR-γ ligand for dopaminergic neurons [107]. In an LPS model of neurodegeneration, improvement in cognition deficits and locomotive functions were observed for LPS-challenged mice treated with PEALut providing a positive correlation between PEALut concentration and attenuation of LPS-induced inflammation as well as neuroprotection [108]. Moreover, PEALut decreased neurodegeneration in the MPTP mouse model of PD [109–111]. Clinical evaluations of co-ultra PEALut as a dietary supplement for the treatment of traumatic brain injury patients are underway (NCT03818451). However, limitations associated with translation from preclinical studies remain oral absorption of PEA and an inverse relationship of bioavailability and increased particle size, highlighting the need for uniform micronization to improve bioavailability [112]. Taken together, strategies that engage anti-inflammatory/anti-oxidant pathways remain one of the more examined, yet controversial approaches investigated in PD, and will require further mechanistic evaluation to accommodate an optimal modality capable of efficacious neuroinflammatory attenuation with consequent beneficial outcomes to disease progression.
ADAPTIVE IMMUNITY IN PARKINSON’S DISEASE
Immune cell profiles in PD patients are altered as demonstrated by decreased levels of naive CD4+ and CD8+ T cells with increased central memory CD4+ T cells and Th1, Th2, and Th17 Teffs as well as increased IFN-γ-producing CD8+ T cells with several lymphocyte subsets linked to clinical motor severity [113]. Numbers of naive B cells are decreased, while non-switched memory B cells, double-negative B cells, and CD19+ B cells that produce TNF-α are increased. While studies enticingly implicate a skewed inflammatory immune system in PD patients, contrary evidence suggest decreased CD8+ Teffs, lower NK cell function, and activated monocytes are also associated with PD [114]. Meta-analysis of 21 case-control studies that included 943 PD patients confirmed the decreased levels of CD3+ and CD4+ T cells in peripheral blood compared to controls, while numbers of NK cells were increased [115]. Levels of CD8+ T cells were also diminished, but only among those in intermediate to late stages of PD. Moreover, this study confirmed a large variability in T cell subsets between studies. It is thought that considerable variation of immune phenotypic and functional profiles may be due, in part, to accompanying stages of disease progression or immunosenescence associated with age-related dysregulation [116]. However, meta-analyses did not confirm age-associated effects compared to age-matched controls, but rather considered inter-study experimental error as a source of variation [115]. Regardless of details on peripheral immunity in PD, these clinical results taken together, suggest a pattern of systemic and adaptive immune dysregulation in PD [114]. Therefore, prospective immunotherapies targeting dysregulated adaptive immunity in PD patients can transform pro-inflammatory to anti-inflammatory and/or reparative profiles, which may improve clinical outcomes that extend beyond the palliative standard of care currently offered [117–119] (Fig. 1).
DMT FOR EFFECTOR T CELLS (Teffs)
After exposure to nitrated α-syn (N-α-syn), Teffs can recognize the modified self-antigen (N-α-syn), and break or evade immunological tolerance and perpetuate disease [120]. Once Teffs expand in the periphery, they migrate and extravasate to areas of tissue damage (Fig. 2). This process is furthered by neuroinflammation with production of pro-inflammatory cytokines and chemokines that include, but are not limited to CXCL12, IL-1, TNF-α, and IFN-γ [121–123]. These factors induce the BBB endothelium to upregulate selectins and adhesion molecules recognized by cognate integrins expressed by Teffs. Endothelium adherent Teffs passage across the BBB to foci of inflamed microglia, are reactivated upon recognition of cognate antigen peptide-MHC presented by microglia, and then elicit interactive immune responses with other microglia to generate a cyclic cascade of exacerbated inflammation, protein modification, and neurodegeneration that perpetuate disease progression [120, 124–127]. Notably, α-syn reactive Teffs induce antigen-specific injuries within an otherwise tolerogenic microenvironment [118]. In support of this idea, we have shown that adoptively transferred N-α-syn-specific Th1 and Th17 Teffs, which produce IFN-γ, TNF-α, and IL-17a, exacerbate MPTP-induced neuroinflammation and dopaminergic neurodegeneration [120, 125]. In those studies, N-α-syn-specific Th17 cells exhibited 50% greater neurotoxicity than Th1 cells. Underscoring these studies is the association of α-syn-specific Th1, Th2, and Th17 CD4+ Teffs as well as cytotoxic CD8+ T cells from the peripheral blood of PD patients and prodromal subjects that respond to in vitro challenge with α-syn-derived peptides [117–119, 128]. One plausible strategy is to inhibit proliferation of pro-inflammatory cells with immunosuppressive agents. Azathioprine is an immunosuppressant that inhibits synthesis of purine nucleotides, DNA synthesis, and thus, proliferation of highly active cells such as Teffs, activated B cells, and myeloid cells such as reactive microglia [129–132]. To test whether suppression of immune proliferation in PD can slow disease progression, a phase II randomized, placebo-controlled, double-blind clinical trial was initiated with daily doses of 1 mg/kg azathioprine and escalating doses to 2 mg/kg (ISRCTN14616801) [133]. The trial is currently ongoing and is scheduled for 12-month treatment period and 6-month follow-up with primary outcome of selected sections of the MDS-UPDRS part III.
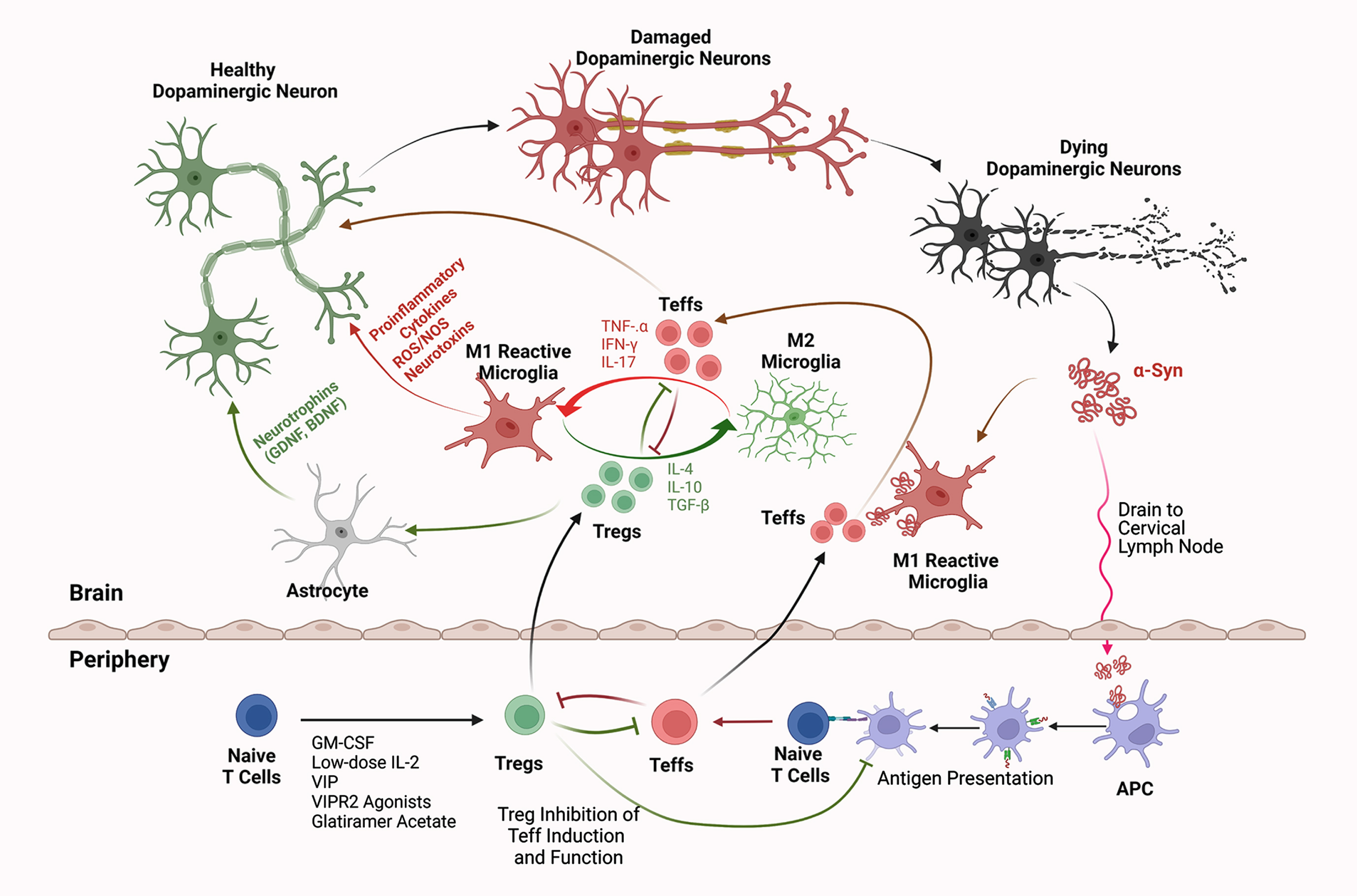
Interactions of Innate and Adaptive Immunity in Parkinson’s Disease. Intraneuronal aberrantly folded α-syn is liberated from injured or dying dopaminergic neurons. Consequent microglial activation leads to a secretory neurotoxic pro-inflammatory cascade within the substantia nigra pars compacta. Misfolded and modified α-syn drains into secondary lymphoid tissues where the adaptive immune system is engaged. This occurs, in measure, by antigen-presenting dendritic cells (APCs) which present modified self-antigens that include nitrated-α-syn, to naïve T cells and elicit Teffs such as Th1, Th2, and Th17 Teffs. Numbers of polarized Teffs increase by clonal expansion. In areas of focal inflammation in the brain, chemokine gradients form and promote endothelial cell to upregulate adhesion and selectin molecules that are recognized by cognate integrin molecules upregulated by expanded and circulating Teffs. Teffs cross the BBB and are reactivated by microglia, which further exacerbate neuroinflammation and neurodegeneration through secretion of TNF-α, IFN-γ, and IL-17. In contrast, Treg-mediated immunosuppressive therapies increase Treg numbers or function with production of anti-inflammatory or immunosuppressive factors, such as IL-10, IL-4, and TGF-β. This promotes a neurotrophic environment by suppressing Teff activation, proliferation, secretion of neurotoxic mediators, and brain infiltration. Additionally, Tregs that cross the BBB suppress microglial and APC activation, attenuate inflammation, block Teff reactivation and cytokine secretion, induce astrocytic neurotrophins, and protect or rescue dopaminergic neurons from further damage. Thus, therapies that induce and/or expand Treg number or function can transform neurotoxic and neurodegenerative conditions into more neurotrophic microenvironments. Created with BioRender.
REGULATORY T CELLS (Tregs) AS DMT
Another strategy to harness uncontrolled innate and adaptive immunity is to potentiate Tregs. Tregs are a CD4+ T cell subpopulation that play critical roles in the maintenance of immunological homeostasis and the limitation of pathogenic defense mechanisms to restrict responses that might lead to immune-mediated pathologies. Tregs are responsible for maintaining active immune tolerance and exert their immunomodulatory activities through several mechanisms that include suppressive and anti-inflammatory cytokines, IL-10 and transforming growth factor-β (TGF-β). Tregs also induce cytolysis of hyper-reactivate inflammatory cells, suppress antigen presentation by dendritic cells, induce T-cell anergy in naïve T cells, alter Teff cell metabolic responses, and serve as a sink for T cell growth factors such as IL-2 [125, 134–136]. Some Treg subsets can bind sufficient levels of IL-2 that preclude Teff cells from utilizing it for clonal expansion, ultimately restricting their expansion. Thus, Tregs serve to control Teff-associated neurotoxicity (Fig. 2). Based on Treg functions, therapies have emerged which increase the number or function of Tregs (Fig. 1) [137]. For many inflammatory–mediated autoimmune diseases, Treg numbers or function are substantially diminished. Therefore, therapeutic strategies would serve to restore those numbers and function to an appreciable level to harness uncontrolled inflammatory response. For the CNS, homeostasis could be achieved by shifting pro-inflammatory neurotoxic responses to anti-inflammatory neurotrophic environments while promoting immune tolerance. Underscoring the importance of Treg activity in harnessing disease and maintaining tissue homeostasis is demonstrated with the loss of Tregs and Treg activity resulting in subsequent loss of immune tolerance. This typically results in a multi-organ systemic inflammatory autoimmune disorder known as immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome, which is fatal in the absence of a bone marrow transplant or Treg reconstitution [138]. When immune tolerance is evaded or broken in neurodegenerative disorders, the host immune system more readily recognizes self or modified-self antigens, such as oxidatively modified or misfolded α-syn, as foreign (Fig. 2). Deficits in Tregs and Treg functions have been demonstrated in several autoimmune disorders such as multiple sclerosis, type-1 diabetes, and inflammatory bowel disease [139].
We and others have shown that PD patients exhibit reduced Treg numbers and/or function as well as increased numbers of Teffs compared to age- and environment-matched caregiver controls [55, 141]. Additionally, evidence that modified α-syn can break or evade tolerance has been demonstrated in pre-clinical models. For instance, immunization of mice with N-α-syn reduces Treg function and induces N-α-syn specific Teffs [125] (Fig. 2). In the MPTP model, within hours of intoxication, N-α-syn and an inflammatory milieu from the CNS drain to cervical lymph nodes and activate antigen presenting cells (APCs) that, in turn, engage the adaptive immune system to elicit antibodies which recognize N-α-syn, but not unmodified α-syn [120]. Additionally, adoptive transfer of Tregs to MPTP-intoxicated animals is sufficient to restore immunological homeostasis, attenuate neuroinflammation, and provide protection for dopaminergic neurons in the substantia nigra and striatum [125]. Interestingly, co-transfer of the same number of Tregs with N-α-syn specific Th17 or Th1 Teffs yields greater Treg-mediated protection, however, greater protection is attained with more inflammatory and neurotoxic Th17 cells than with Th1 cells. This suggests that the transformative efficiency for Tregs is somewhat dynamic and depends on the conditions requiring immune regulation. Moreover, Tregs have been shown to be most efficient at controlling neurotoxic microglia activated and/or augmented by extraneuronal N-α-syn [120, 143]. Taken together, Tregs attenuate microgliosis, spare dopaminergic neurons from death, rescue striatal termini, and downregulate pro-inflammatory mediators, such as iNOS, TNF-α, and IL-1β [144, 145] (Fig. 2).
Immunoregulatory peptides and growth factors, such as vasoactive intestinal peptide (VIP) and granulocyte-macrophage colony-stimulating factor (GM-CSF) are immunomodulatory agents that induce or augment Treg numbers and/or activity as measured by the ability to inhibit CD3/CD28-stimulated conventional T cells, designated as responder T cells (Tresps) [56, 146–148] (Fig. 1). Administration of either agent before or at the time of neuronal insult in MPTP or α-syn over-expression models increases Treg function, attenuates microglial activation and can protect 100% of dopaminergic neurons eliminated due to neurodegeneration [146, 149] (Fig. 2). However, both agents exhibit relatively short half-lives that present limitations for their clinical use. To overcome the short half-life of VIP, a VIP receptor-2 (VIPR2) peptide agonist, LBT-3627, was developed as a stable immunomodulatory agent with a 24-fold increase in half-life, but with 10-fold less potency [148, 149]. After five daily doses of LBT-3627, Treg function was increased compared to vehicle control [149]. In the mouse MPTP model and in the rat 6-hydroxydopamine (6-OHDA) and α-syn overexpression models, treatment with LBT-3627 or adoptive transfer LBT-3627-induced Tregs, reduced numbers of reactive microglia and improved dopaminergic neuron survival [148–150]. This agent is currently being developed for potential therapeutic use in PD patients.
In preclinical studies, treatment with GM-CSF also attenuates reactive microglia, induces robust anti-inflammatory shifts from Teff-dominant environments, and preserves significant numbers of dopaminergic neurons in the MPTP or α-syn overexpression models [56, 147]. It also induces significant increases in Treg numbers in a dose-dependent fashion, and adoptive transfer of those Tregs spare nigral dopaminergic neurons. In a double-blind, placebo-controlled phase Ia clinical trial, 6μg/kg/day of human recombinant GM-CSF (sargramostim, Leukine®) administered for eight weeks was found safe and well-tolerated [151]. By one week after initiation of sargramostim treatment, Treg numbers were increased over placebo controls during the duration of treatment interval, and the mean Treg activity was significantly elevated over that of controls and entry baseline. By six weeks after initiation, UPDRS, part III scores showed modest, but significant improvements compared to controls and baseline, which returned to baseline after drug discontinuation. At study entry, magnetoencephalography (MEG) showed decreased cortical activity in areas of motor control, which significantly increased after eight weeks of sargramostim treatment. While deemed safe and tolerable, mild side effects were observed that included injection site reactions, bone and limb pain, and elevated white blood cell counts. This led to an open-label 24-month phase Ib clinical trial with dose reduction to 3μg/kg/day of sargramostim in a five-day on/two-day off regimen. One-year results indicated that monthly adverse events per subject were reduced by 82% compared to those reported in the initial phase Ia trial [152]. In parallel, Treg numbers were increased by one month of treatment, and by two months, Treg activity was increased and remained above entry baseline activity during the course of treatment. Similar to the phase Ia trial, these results paralleled improvements in UPDRS part III scores. While other Treg-based strategies have been used, including adoptive transfer of expanded autologous Tregs in other neurodegenerative disorders, such as amyotrophic lateral sclerosis, the dysfunctions of the endogenous Treg source pool may be difficult to overcome and may affect post-administration survival and activity, thus rendering diminished capacities for sustained clinical benefit leading to continued disease progression after transfer [153]. Therefore, it may be necessary to repopulate Tregs from bone marrow progenitors and expand those new Tregs to obtain stable, functional Tregs. We recently extended this concept by showing significant neuroprotective capacity of Tregs that were expanded in vivo by low-dose IL-2, and adoptively transferred to MPTP-treated recipients [154]. Ongoing strategies to increase Treg numbers and function have focused on extending the six-hour half-life of GM-CSF. Such increases, if possible, would overcome limitations in the overall dose, dose frequency, bioavailability, and potential adverse events for its long-term utilization. To such ends, we developed formulations with longer half-lives and increased bioavailability allowing for decreased frequency of drug administration with increased Treg-mediated function and dopaminergic neuroprotection [56, 155]. Other immunomodulatory strategies explored at the preclinical level include, but are not limited to, histone deacetylase inhibitors, corticosteroids, bee venom phospholipase A2, CD3 antibodies, Bacillus Calmette–Guérin vaccine, low-dose IL-2, and combined TGF-β with IL-10 [156–158]. These possess parallel neuroprotective activities [159, 160]. Together, the search for a safe and effective DMT for immune transformation in PD that restores homeostatic microenvironments to the inflammatory brain and potentiates neuronal survival and function, remains an active area of investigation [161].
While Tregs that exhibit indeterminate specificity seem to provide adequate protection, they may not restore long-term antigen-specific immune tolerance. One strategy could isolate antigen-specific Tregs by sorting and expand those for adoptive transfer to directly re-establish immunological tolerance. However, this would require identification of the cognate antigen peptide and synthesis of peptide/MHC conjugates by using tetrameric MHC molecules or multimeric MHC molecules on a dextran backbone (dextramer) for the isolation of antigen-specific Tregs. Moreover, patient-specific MHC compatibility would be needed to ensure appropriate recognition of the peptide/MHC by the T cell receptor (TCR) target cells and would ultimately require independent quality control measures for each subject isolate. Once isolated, antigen-specific Tregs would need to be expanded and validated for the capacity to recognize the peptide/MHC complex and may need to show in vitro suppressive function [162]. Thus, the required steps necessary to obtain an efficacious antigen-specific, MHC-restricted therapeutic Treg modality may limit its general utilization in neurodegenerative disorders. However, a recent study suggests that MHC restriction may not be required for Treg therapeutic efficacy [163]. In a model of experimental colitis in mice with a reconstituted human immune system from 4 unique HLA-mismatched donors, low-dose IL-2 expanded Tregs protected mice from colitis compared to controls. Thus, without the necessity of MHC compatibility, Treg replacement from allogeneic sources would provide a strategy for a safe, readily available, and reproducible method to establish immunological tolerance to altered self-antigen. Indeed, targeting Treg functional restoration is preferable as these cells mitigate neuroinflammation and induce neurotrophic support, which attenuate neuronal loss and slow protein modification and misfolding during disease [56, 152].
Bypassing the requirement for TCR recognition of peptide-MHC could be accomplished by engineering Tregs that express the peptide antigen recognition sites of antibodies, thus constructing a chimeric antigen receptor (CAR)-Treg targeted to recognize modified self-antigens [164] (Fig. 1). As with other CAR T-cell platforms, immunoglobulins provide the primary antigen recognition apparatus directed at modified moieties on pathogenic self-proteins, such as nitrated-tyrosine or phosphorylated-serine of α-syn. Genes that encode a single-chain containing the heavy and light chain antigen-binding fragments (scFv region) would be transduced into either autologous or allogeneic Tregs to produce CAR-Tregs. Indeed, CAR-Tregs have been constructed in preclinical models to recognize modified self-antigens in autoimmunity, allo- or xeno-antigens in transplantation whereby immune dysregulation results in cell- or humoral-mediated hyper-responsivity [165]. For PD and other inflammation-mediated neurodegenerative diseases, CAR-Tregs that detect antigen in an MHC-independent fashion would be less dependent on MHC compatibility to maintain stable phenotypes and function. The latter was most recently confirmed using adoptively transferred CAR-T cells as a leukemia therapy, in which leukemia-specific CAR-T cells are detectable and functional ten years post-administration [166]. Similarly, CAR-Tregs would retain the capacity for preferential migration to the inflammatory site and exert a more potent immunosuppressive potential than other Tregs, thus may more efficiently restore non-MHC directed immune tolerance to specific antigens, such as modified α-syn. However, issues associated with CAR T cells in neoplastic disease include initial cytokine storm and peripheral neurotoxicity [166]. Both should be considered with CAR-Tregs, however given the differential opposition of efferent functions and cytokine repertoire of CAR-T cells and CAR-Tregs, issues of cytokine storms and neurotoxicity using CAR-Tregs may be less likely. Lastly, the possibility of Treg exhaustion may affect long-term efficiency of immune tolerance. However, in humanized mice, complete and long-term suppression of HLA-A2 allograft rejection was achieved with CAR-Tregs expressing HLA-A2 specific single-chain variable fragments, whereas polyclonal antigen-specific Tregs could not prevent alloreactive rejection [167]. Overall, Treg-mediated attenuation of neuroinflammation may provide an important role in interventional therapy of neurodegeneration. In PD, immunomodulatory agents, poly- or mono-clonal Tregs, or CAR-Tregs could impart a therapeutic bridge to inhibit further loss of dopaminergic neurons. Moreover, for therapies targeting neuronal replacement, Treg-mediated therapies may be required to harness neuroinflammation and ameliorate synucleinopathy to prevent subsequent neurodegeneration and loss of transplants [33, 34].
DMT FOR α-SYNUCLEIN
Misfolding and aggregation of α-syn induce oxidative stress, autophagy and synaptic dysfunction, protein ubiquitinylation, and eventual LB formation [20, 168]. An inflammatory microenvironment of the midbrain perpetuates the formation of modified α-syn and ubiquitin proteins. From these processes, CNS proteins drain from the brain to peripheral lymphoid tissues, such as deep cervical lymph nodes, and engage APCs (Fig. 2). APCs release pro-inflammatory signals and present modified self-antigens, like N-α-syn, to naïve T cells to induce Teff immune responses [143]. Through pro-inflammatory co-stimulatory signals, naïve CD4+ T cells can be programmed into Teffs that are polarized by secondary and tertiary co-stimulatory signals. Under most circumstances, the presentation of self-antigen fails to produce pathogenic autoreactive Teffs; however, modified self-antigen can elicit pathogenic Teffs [117]. This includes the emergence of autoreactive T cells as demonstrated in PD and PD models [120, 125]. Conditions for adaptive immune recognition of modified N-α-syn have been described, whereby within hours of MPTP-intoxication, N-α-syn and activated APCs are found in cervical lymph nodes with consequent T cell and antibody responses to N-α-syn, but not α-syn, acquired days later [120]. Spreading of aggregated oligomeric α-syn species also elicits systemic neuroinflammatory responses due to intraneuronal toxicity of its fibrils and production of neuronal-derived damage-associated molecule patterns [118, 169]. These are secreted or released into the extraneuronal spaces upon neuronal injury or death and can potentially activate microglia to high states of reactivity and induce autoreactive T cell responses. Thus, therapeutic strategies that target α-syn clearance would prevent spreading of aggregated α-syn oligomers and eliminate its extended availability to perpetuate inflammatory microglia. Moreover, its elimination would preclude interaction, processing, and presentation by APCs, and lessen the chance for afferent T cell induction. Therefore, clearing this potent activator of the immune response would restore a more tolerogenic, anti-inflammatory, and neurotrophic environment by disrupting the autoreactive T cell- and microglia-mediated neural damage.
One strategy is to clear oligomeric and aggregated α-syn with anti-α-syn antibodies either produced by active immunization (i.e., directly immunizing the host) or by passive immunization (i.e., parenteral administration of antibodies) (Fig. 1). Several trials have been reported using either monoclonal antibodies (mAbs) or vaccines for α-syn clearance. Cinpanemab (BIIB054), was created as an IgG1 anti-α-syn antibody derived from memory B cells of elderly individuals with CNS diseases [170]. BIIB054 binds the N-terminal aa1-10 residues and demonstrates an 800-fold higher affinity for α-syn. In mice that were intracerebrally administered preformed α-syn fibrils, BIIB054 inhibited α-syn spread and aggregation, diminished loss of striatal dopaminergic termini, and rescued motor impairments. In a phase I trial of PD patients, BIIB054 displayed a half-life of 28–35 days and almost complete saturation of the bioavailable BIIB054 with α-syn [171]. Ultimately, a phase II study was terminated due to failure to meet endpoints in baseline UPDRS, Parts I, II, and III score at weeks 52 and 72. A second monoclonal antibody to α-syn, ABVV-0805, preferentially recognizes aggregated α-syn and demonstrated dose-dependent reduction in α-syn aggregates in several mouse models and increased survival in PFF-treated A30P-α-syn transgenic mice [172]. However, a phase I trial was withdrawn before subject enrollment due to “strategic considerations” (NCT04127695). Lastly, PRX002/RG7935 (PRX002, prasinezumab) is a humanized IgG1 mAb that recognizes the C-terminus of α-syn and targets aggregated forms of α-syn, thus reducing α-syn intercellular propagation, neuropathology, and behavior deficits in a PD-like model [173, 174]. In a phase I trial, PX002 was deemed safe and tolerable, and showed vigorous binding of peripheral α-syn with dose-dependent increases of mAb detected in cerebrospinal fluid; thus it was deemed likely to engage extracellular aggregated α-syn in the brain [175]. In a year-long phase II trial of early-stage PD patients [176], PRX002 did not lessen progression of motor and non-motor symptoms in part 1 of the study compared to placebo controls, but it did show signs of efficacy in secondary and exploratory measures (NCT03100149). In part 2 of the study, trends affecting progression of motor severity were reported. This subset of PRX002-treated subjects were in an early start group that were either drug naïve or also treated with monoamine oxidase B inhibitors, and exhibited diminished UPDRS, part III scores compared to a delayed start group. Based on those findings, a phase IIb clinical trial has now been initiated for subjects with more advanced symptoms to further evaluate the efficacy of PRX002 (NCT04777331) [177].
Active vaccination using α-syn or α-syn mimetics represent another strategy to clear α-synuclein from the brain. Two leading α-syn mimetic vaccines, PD01A and PD03A, are in development [178]. These are composed of short peptides of eight and ten amino acids, respectively, and were selected from a library of synthetic peptides that recognize and bind antibody directed against α-syn oligomers [179]. Both mimic epitopes on the C-terminus of α-syn and target pathogenic assemblies of the protein. Preclinical studies have established tangible neuroprotective benefits with decreased accumulation of α-syn species. As vaccine candidates, peptides were conjugated to keyhole limpet hemocyanin (KLH) as a T cell carrier protein and adjuvanted with aluminum hydroxide. In phase I trials, PD subjects were immunized with four 15 or 75μg doses [178, 180]. Both vaccines were found to be safe and tolerable and increased antibody titers to α-syn. PD01A generated antibody titers in 89% of the subjects, whereas the responder rate from those receiving PD03A was 58%, and titers were lower than those associated with PD01A. An additional PD vaccine candidate, UB-312, is another synthetic ten amino acid peptide from the C-terminus of α-syn and was selected from sixty B cell α-syn epitopes that showed increased immunoreactivity in guinea pigs [181]. UB-312 was adjuvanted with polyanionic cytosine phosphoguanine (CpG) oligodeoxynucleotide and an aluminum phosphate gel (Adju-Phos®). In an α-syn transgenic model of PD, UB-312 immunization induced anti-α-syn antibodies, reduced α-syn oligomers in the cerebral cortex, hippocampus, striatum, and colon, reduced reactive microglia, and prevented development of motor impairments [181]. UB-312 is currently in a phase I clinical trial (NCT04075318). Together, reduced α-syn loads and increased neuroprotection in preclinical studies combined with initial results of clinical trials lend support for vaccination strategies that target α-syn to halt its spread and accumulation.
NANOBODIES AS DMT
An alternative immunotherapeutic strategy utilizes nanobodies to destabilize or inhibit intracellular α-syn aggregation and accumulation of toxic fibrils (Fig. 1). Nanobodies are proteins containing only the antigen binding portions of the light (L) and/or heavy (H) chains of the antibody variable region (V), such as the fragment antigen binding (Fab) domains, single chain antigen binding (VL+VH) (scFv) domain, or single-domain Ab (VHH) domain. Therefore, these immunoglobulin-derived domains bind the antigenic epitope to which the antibody was directed. Typically, these proteins are obtained from high affinity antibodies, since efficacy depends on only the small antigen-binding portions of the antibody. Two nanobodies that have been isolated from camelid antibodies, NbSyn2 and NbSyn87, show high degrees of affinity towards distinct epitopes of the α-syn C terminus, aa118–131 and aa137–140, respectively [182–184]. These have been well characterized and assessed for their potential therapeutic utilization [183, 185]. Both nanobodies selectively and efficiently bound and inhibited α-syn fibrillation, but their affinities varied significantly depending on the length of time during α-syn fibrillation and the state of the fibrils. Two scFv nanobodies (NAC32 and VH14) that recognize aa53–87 of the α-syn non-amyloid component (NAC) region were developed, and the coding sequences incorporated into adeno-associated virus (AAV) constructs to be used as intracellularly expressed nanobodies, termed intrabodies [186]. Co-transfection of each construct with AAV-αsyn constructs into HEK293 and SH-SY5Y cells demonstrated that co-expression of α-syn and NAC32, but not VH14 intrabodies, downregulated α-syn protein, but not α-syn mRNA [187]. Co-administration of AAV-NAC32 with AAV-αsyn to rats, reduced α-syn expression in the substantia nigra and striatum, and spared dopaminergic termini in the striatum. In a rat model of DAT-Cre selective expression of α-syn, nigral injection of AAV-NAC32 diminished α-syn expression, spared nigral dopaminergic neurons, and improved locomotor measurements in open-field monitoring [188]. Intrabody susceptibility to poor intracellular solubility and a propensity to aggregate can potentially increase the protein burden and neurotoxicity associated with intrabody expression, such as with VH14. Thus, delivery of genetic constructs that are translated to yield therapeutic intrabodies, must evade aggregation as well as intracellular degradation. To enhance this evasion, intrabody constructs have been engineered with a polypeptide rich in proline, aspartate/glutamate, serine, and threonine (PEST) residues to increase net charge and isoelectric point, and to improve intracytoplasmic expression levels [182]. Additionally, PEST residues act as ubiquitin-independent proteasome-targeting motifs that can chaperone intrabody-bond antigens to proteasomal degradation. To test the effect of PEST modifications to inhibit intracellular α-syn accumulation, two nanobody constructs, VH14 and NbSyn87 were modified to include the PEST motif for expression as intrabodies, and were delivered by stereotaxic injection to α-syn overexpressing rats [182]. Compared to controls, both intrabody*PEST constructs diminished phosphorylated α-syn, and while VH14*PEST showed increased rescue of dopaminergic striatal termini, levels of dopamine transporter immunoreactivity, and dopamine neurotransmitter levels, increased numbers of reactive microglia were detected after treatment with NbSyn87*PEST constructs. Thus, these data underscore the utility of PEST modifications to transform non-clearing intrabodies, such as unmodified VH14, to a species capable of clearing accumulated α-syn to control levels [182, 187].
While clinical trials have yet to include intrabody constructs as delivery platforms directed against α-syn, other nanobodies were developed to be delivered as extracellular biologics and have entered or completed clinical trials, including those with specificities to IL-17A/F (bimekizumab (ALX-0761/MSB0010841), NCT02156466) [189]; IL-17A/IL-17F/human serum albumin (sonelokimab (M1095), NCT03384745) [190, 191]; IL-6 receptor (vobarilizumab (ALX-0061), NCT01284569) [192]; CXCR4 (ALX-0651, NCT01374503); A Disintegrin And Metalloproteinase with ThromboSpondin motif (ADAMTS)-5 (M6495, NCT03224702 and NCT03583346) [193]; von Wiillebrand factor (caplacizumab (ALX-0081), NCT01151423 and NCT02553317) [194]; respiratory syncytial virus (gontivimab (ALX-0171), NCT01483911) [195]; human serum albumin VHH nanobody-IL-21 (JS014, NCT05296772); and PD-L1 (68Ga-THP-APN09, NCT05156515). With α-syn-directed vaccines and therapies now at hand, abundant preclinical findings reinforce the capacity of nanobodies to inhibit or destabilize α-syn aggregation and fibril formation, which would decrease α-syn loads and provide support for their utilization in PD.
GENE THERAPY AS DMT
With the advent of gene editing technology, gene therapy is becoming increasingly more reliable, accurate, and effective. The premise of gene therapy is to replace, alter, or edit deleterious gene expression responsible for disease occurrence and activity (Fig. 1). Strategies implementing gene therapy for PD have largely investigated the possibility of enhancing a sustained release of dopamine from damaged dopaminergic neurons [196]. While these strategies warrant further attention and resources, pursuing methods that seek to address the genetic complexity responsible for immune imbalance may prove to be beneficial. One strategy targets a family of genes that collectively initiate or enhance transcription of the anti-inflammatory cytokine, TGF-β. Among these candidate genes is glial cell-derived neurotrophic factor (GDNF) (Fig. 1), which has been shown in many preclinical studies to attenuate neuroinflammation and oxidative stress, and provide neuroprotection [197–202] as well as stimulate and regenerate dopaminergic neurons [203]. Possible mechanisms for GDNF-mediated effects on microglia include activation of the GDNF family co-receptor α1 (GFRα1) and the transmembrane receptor tyrosine kinase (RET) complex, inhibition of the focal adhesion kinase (FAK) pathway, and reduction in p38 phosphorylation that leads to diminished p38-MAPK signal transduction [197, 198]. Moreover, our laboratories have shown that adoptive transfer of Tregs to MPTP-treated mice not only attenuates neuroinflammation and protects dopaminergic neurons, but also upregulates GDNF and BDNF expression by astrocytes [204]. An early phase I trial delivering GDNF via intracerebroventricular injection yielded no improvement in UPDRS scores and produced several adverse events (NCT00006488). However, in a second phase I trial, GDNF was delivered via intraparenchymal catheters and resulted in 30–40% improvement in UPDRS scores, increased uptake of 18F-dopamine by dopaminergic neurons, and reduced dyskinesia with few adverse events [205]. However, after a phase II trial demonstrated no significant improvement in UPDRS scores [206] and contemporaneous studies in non-human primates showed loss of Purkinje cells and cortical pathology [207], the sponsor terminated the program. Nevertheless, due in part to phase I results, GDNF has remained a therapeutic strategy for many researchers as well as PD patient advocates [203], and has led to intense efforts to find a better delivery strategy for GDNF. One approach is to deliver the GDNF gene in an AAV construct. In an open-label phase I study, the gene for neurturin (NRTN, a GDNF homolog) was delivered via bilateral intraputamenal injections as an AAV2-NRTN construct (CERE-120) (NCT00252850). The treatment was well-tolerated over the one-year study without evidence of dyskinesia and improved several motor function sub-scores of the UPDRS measures from baseline, while other secondary measures were not significantly affected nor were changes in t8F-levadopa uptake detected [208]. A phase Ib study is currently underway to test intraputamen injection of AAV2-GDNF in PD for safety with the rationale that such delivery can better recapitulate dopaminergic neuroprotection observed in preclinical studies [209, 210].
DMT WITHIN THE GUT-BRAIN AXIS
The gut-brain axis (GBA) is a prospective area of research, which has generated a new appreciation for the symbiotic relationship that governs neurochemistry, behavior, and host neuroinflammatory environment [211, 212] (Fig. 1). The GBA consists of cross-talk networks between the microbiome in the gut, peripheral immune system, and the CNS. The microbiota composition of the gastrointestinal (GI) tract has been found to uniquely influence not only the peripheral immune environment, but also CNS microglia activity as well [213–216]. Moreover, a potential connection exists between early GI dysfunction and later developing stages of PD [217, 218]. In support of this, is a growing abundance of evidence implicating the role of intestinal dysbiosis in disease progression [219]. This is congruent with the Braak staging hypothesis, which posits that initiation of PD transpires from early misfolding of α-syn in the olfactory system or gut, and spreads via the olfactory tract or the vagal nerve to the CNS, and that the microbiome may control early oligomerization of α-syn [220, 221]. Thus, one strategy would selectively alter the commensal microbial community, providing an avenue towards addressing the associations of the gut microbiome with peripheral and central immune dysfunction in PD. While one strategy targets several combinations of microbes to provide the optimal microbiome, a few probiotics have been discovered that possess neuroprotective capabilities and are currently in clinical trials [222–224]. Of the probiotics under investigation for PD, the two most commonly reported genera of microorganisms used as probiotics in clinical trials are Lactobacillus spp. and Bifidobacterium spp. [225]. In PD mouse models, a significant improvement in motor function and dopaminergic preservation was reported in probiotic-treated mice containing both Lactobacillus and Bifidobacterium spp. [226]. Considerable numbers of trials will be required to uncover a defined microbiome that can colonize the gut with sufficient retention to provide significant beneficial alteration of the processes that would diminish neuroinflammation, mitigate neurodegeneration, and/or provide neurotrophic support for dopaminergic neuronal survival. With over 15 clinical studies underway focusing on the microbiome, the importance of the gut-brain axis, not only in PD, but also for other neurodegenerative disorders, underscores its potential impact [227].
CONCLUSIONS
Evidence from preclinical studies and clinical trials suggests that modulation of immune responses provide treatment options as DMTs that could extend beyond current palliative management, and tangibly alter the processes that underlie neurodegenerative mechanisms in PD. However, to utilize these options in a therapeutic fashion requires a greater understanding of molecular mechanisms associated with PD onset, roles of the immune system in neuroinflammation, and subsequent processes leading to preferential neurodegeneration along the dopaminergic nigrostriatal axis. Nonetheless, therapies that target detrimental innate inflammatory responses initiated or potentiated by autoreactive T cells will generate new pharmacologic avenues. The lack of translational potential for many anti-inflammatory agents that do not meet primary or secondary endpoint measures in clinical trials underscores the complexity of these neurodegenerative processes, particularly in PD. While negative outcomes of clinical trials emphasize the difficulty in obtaining an effect in PD populations for a variety of reasons, those trials along with the few others that yield beneficial outcomes, should serve to bolster DMT interventions. For instance, inflammation and reactive oxygen species accumulation play a clear role in initiation and/or acceleration of cell death in PD, yet numerous large and well-conducted clinical trials targeting oxidative stress and inflammation have shown little improvement in clinical outcomes [228]. The lack of objective and accurate measures of dopaminergic loss or inhibition of loss limits the validity of the trials as only indirect and subjective tests are available for clinical outcomes. Also, imaging may not correlate with dopamine neuron density, and while most PD subjects are enrolled within five years of diagnosis, approximately 50–70% of nigral dopaminergic neurons may already be lost, thus leaving an insufficient level of functional neurons to detect an effect with the tools available [11]. Thus, many top preclinical therapeutic candidates trialed without beneficial outcomes, could have plausibly produced positive outcomes, but went undetected due to limits of experimental resolution. Nonetheless, sufficient positive preclinical studies have fueled development and emergence of anti-inflammatory agents, such as GM-CSF, VIP agonists, Tregs, or co–ultra PEALut [62, 230]. Thus, alternative combinatorial approaches that utilize non-traditional modalities, such as Treg-based therapeutics, gene therapy, or α-syn-targeted approaches may be needed for intervention of the many pathways that govern preferential vulnerability and loss of dopaminergic neurons in PD.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.