
Abstract
Background:
Prodromal multiple system atrophy (MSA) has been characterized mainly by retrospective chart reviews. Direct observation and tracking of prodromal markers in MSA have been very limited
Objective:
To report the baseline characteristics and evolution of prodromal markers of MSA as they were prospectively measured in patients with idiopathic/isolated REM sleep behavior disorder (iRBD)
Methods:
Patients with iRBD were evaluated as part of a comprehensive protocol repeated annually. The protocol included assessment of motor, sleep, psychiatric, and autonomic symptoms supplemented by motor examination, quantitative motor testing, neuropsychological examination, orthostatic blood pressure measurement, and tests of olfaction and color vision. Patients who eventually developed MSA were described and compared with those who phenoconverted to Lewy body disease (Parkinson’s disease and dementia with Lewy bodies).
Results:
Of 67 phenocoverters, 4 developed MSA-P and 63 developed Lewy body disease. An additional 2 MSA-C patients were seen at baseline, already with cerebellar signs. Compared to those with Lewy body disease, those with MSA-P were younger, had less severe loss of tonic REM sleep atonia, more insomnia symptoms, and better olfaction. Clinically-evident autonomic dysfunction was not invariable in prodromal stages, often developing proximate to or after motor phenoconversion. Of the autonomic symptoms, genitourinary dysfunction was the first to develop in all cases. Olfaction and cognition remained normal throughout the prodromal and clinical disease course, in clear contrast to patients with Lewy body disease.
Conclusion:
Prodromal MSA progresses rapidly, often without substantial autonomic dysfunction, and with preserved olfaction and cognition throughout its prodromal course.
Keywords
INTRODUCTION
Multiple system atrophy (MSA), like all neurodegenerative conditions, has a prodromal stage, namely a period in which some clinical signs or symptoms of disease are evident but are not yet at threshold for clinical diagnosis. The ability to directly measure this stage has been extremely limited, mainly because there are few methods to reliably detect prodromal MSA. This represents a critical unmet need, as it will be crucial to intervene as early as possible with neuroprotective therapy once this becomes available.
Most of what is known about prodromal MSA comes from retrospective recall or chart review of clinical symptoms among those with clinical MSA [1]. Predominant symptoms of the prodromal state include autonomic failure and REM sleep behavior disorder (RBD). Most studies of prodromal MSA have come from autonomic laboratories; studies have suggested that approximately 4–20% of patients diagnosed with pure autonomic failure will develop MSA over the subsequent 4–10 years [2–5]. However, autonomic failure is not the only prodromal presentation of MSA. Large observational studies have found that that approximately 4–5% of patients with idiopathic/isolated RBD (iRBD) will ultimately develop MSA [6], providing a second avenue to directly examine prodromal MSA.
Retrospective surveys of RBD occurrence in MSA have been performed; approximately 30–40% of patients with MSA retrospectively recognize RBD symptoms prior to disease onset, with most of these noting that RBD was the initial symptom [7] (note that many more may have RBD but be unaware). Studies have suggested that the presence of RBD anticipating MSA also predicts poor prognosis, including earlier death [7, 8]. However, studies prospectively tracking prodromal MSA patients from iRBD have been extremely limited. One observational study found that MSA patients had normal olfaction at baseline, in distinction to those who developed Lewy body disease (i.e., Parkinson’s disease (PD) and dementia with Lewy bodies (DLB) [9]. This study only assessed baseline measures and did not describe the evolution over time of prodromal MSA. Over the last 17 years in our center, we have been systematically tracking iRBD patients using a prospective annual follow-up protocol. During this period, we have directly observed 4 patients develop MSA-P from their prodromal state, plus two with MSA-C who already had mild cerebellar findings. We describe here the evolution of clinical and biomarker features of these patients, compare them to those who developed PD and DLB, and discuss the implications of our findings for development of potential prodromal MSA criteria.
METHODS
All patients were enrolled in an ongoing prospective study of iRBD, which has been described in detail elsewhere [10, 11]. Briefly, all patients met criteria for polysomnography (PSG)-confirmed iRBD, defined with standard ICSD-III criteria [12], and were free of parkinsonism or dementia at baseline. Patients were then followed on an annual basis, with a systematic protocol that included motor markers (Unified Parkinson Disease Rating Scale, timed-up and go, alternate tap test, Purdue Pegboard), cognitive evaluation (Montreal Cognitive Assessment and an extensive neuropsychological battery [13]), clinical measures of autonomic function (orthostatic, urinary, erectile, and bowel symptoms on the Unified Multiple System Atrophy Rating Scale [14], systolic blood pressure drop measured manually in the supine and standing position after 1 min), special senses (olfaction with the cross-cultural 12-item version of the University of Pennsylvania Smell Identification Test (UPSIT) test [15], and color discrimination with the Farnsworth-Munsell 100 Hue test (FM-100) [16]), other sleep measures (Insomnia severity Index [17], Epworth sleepiness scale [18], Pittsburgh Sleep Quality Index [19], PSG indices of REM sleep atonia [20, 21]), and depression and anxiety measures (Beck depression and Anxiety Inventories [22, 23]). Other additional diagnostic procedures including neuroimaging [24] and skin biopsy [25] were more recently added to the protocol and assessed on a subset. All participants provided written informed consent and ethics approval was obtained from the local research ethics board.
Patients were then annually assessed with the same protocol and examined for development of defined neurodegenerative syndromes. Outcomes of interest included dementia for parkinsonism (first by UK brain bank [26], then by Movement Disorders Society criteria [27]) or dementia (using DLB criteria [28, 29]). If patients developed parkinsonism, the most likely diagnosis was defined according to best clinical impression, based upon all diagnostic information available at final clinical visit.
RESULTS
Four patients developed MSA-P, all without parkinsonism at baseline examination. Details of each case are provided.
Case descriptions
Please see Table 1 for details of all baseline variables and Fig. 1 for changes over time.
Description of baseline measures in patients who developed MSA-P
MoCA, Montreal Cognitive Assessment; MCI, mild cognitive impairment; UPSIT, University of Pennsylvania Smell Identification Test (12-item cross-cultural version); FM-100, Farnsworth-Munsell 100 Hue test; UPDRS, Unified Parkinson Disease Rating Scale; RBD, REM sleep behavior disorder; MSA, multiple system atrophy.
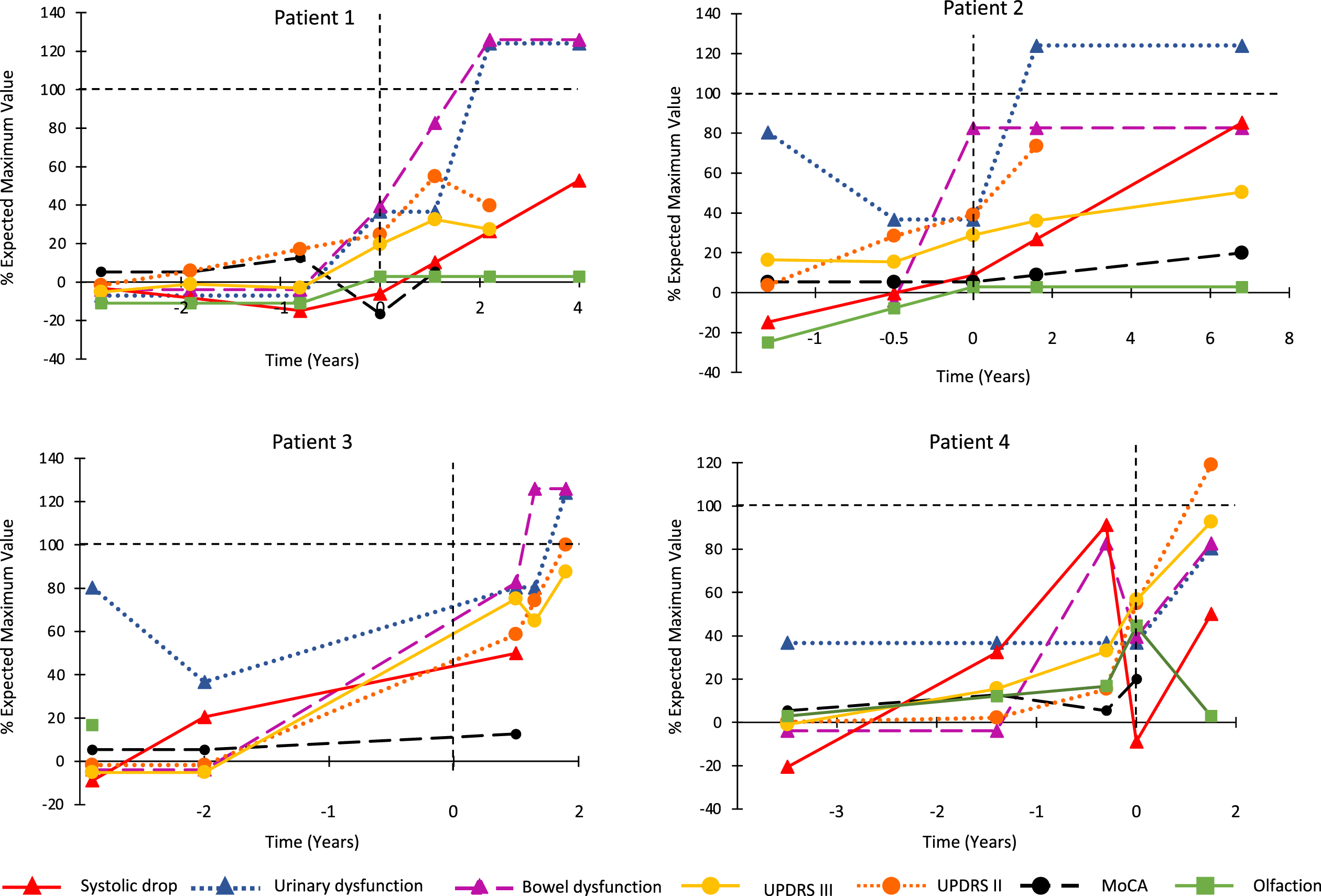
Combined progression trajectory of motor and non-motor manifestations from prodromal stages. For each marker, the 0 point is set as the normal control values, selected from our cohort study. The 100% indicates severe abnormality of the marker, as assessed as the worst 1 standard deviation of patients with advanced Parkinson’s disease and RBD, taken from a previous study [31].
Six months after the previous normal examination, he called to request urgent follow-up for rapid deterioration. Upon this evaluation, UPDRS was now 12, with left sided rigidity and bradykinesia. He now had new erectile dysfunction, urinary frequency with mild retention symptoms, and constipation. There was still no orthostatic hypotension (blood pressure 143/78 lying to 144/78 standing). He received levodopa with clear, but moderate benefit that only became evident at doses of 600 mg per day. Over the next 3 years, he has required high doses of levodopa for symptom control (1500 mg per day) and experienced on-off fluctuations and dyskinesia (dyskinesia include the face). Balance has remained normal. As of last visit, he had evident orthostatic hypotension (systolic drop = 20 mm Hg at 1 min, despite taking fludrocortisone), severe urinary retention requiring catherization (prostate examination was normal), severe bowel dysfunction often requiring manual disimpaction because of sphincter dyssynergia, and complete erectile dysfunction. There was severe bulbar dysfunction, with frequent choking. Exam revealed polyminimyoclonus and brisk reflexes. Cognition and olfaction have remained normal throughout.
MSA-cerebellar
Of note, two cases with previously-undiagnosed possible MSA-C have also been seen at our center. However, in both cases, they did not meet inclusion criteria, because both had mild cerebellar signs on baseline examination. Without defined thresholds for clinically-significant cerebellar dysfunction (unlike is the case for parkinsonism and dementia), these cases cannot be considered to have clearly been prodromal. Therefore, they are not included in this study. However, of note, one of the cases had clear autonomic dysfunction and orthostatic hypotension at baseline (64 mm Hg systolic drop). 5 years later, the patient was wheel-chair bound. The second had clear cerebellar ataxia at baseline with urinary frequency (no retention) and severe erectile dysfunction but had no orthostatic hypotension (with blood pressure measurements at 1 and 3 min standing). One year later, there was clear progression of cerebellar findings, with a new asymptomatic 19-mm Hg systolic drop.
Prodromal MSA versus Lewy body disease
We compared the patients developing MSA-P to 63 iRBD patients who developed Lewy body disease (Table 2). Noting the limited statistical power and exploratory nature of the analysis, we found that the MSA patients at baseline were significantly younger, had less severe loss of tonic REM sleep atonia, more insomnia symptoms, and normal olfaction. In terms of autonomic function, MSA convertors at baseline actually had less orthostatic blood pressure systolic drop and less constipation but had more urinary dysfunction. Point estimates of measures of both cognitive and motor testing suggested better baseline function in MSA, although differences were not significant.
Baseline variables in MSA versus Lewy body disease
p values are calculated using t-test and Fisher exact test. They are presented unadjusted for multiple comparisons; given this, and given the low sample size, they are included for information only, and should be considered exploratory in nature. MoCA, Montreal Cognitive Assessment; MCI, mild cognitive impairment; UPSIT, University of Pennsylvania Smell Identification Test (12-item cross-cultural version); FM-100, Farnsworth-Munsell 100 Hue test; UPDRS, Unified Parkinson Disease Rating Scale; RBD, REM sleep behavior disorder; MSA, multiple system atrophy.
Patterns of evolution are illustrated in Fig. 1, mapped using methodology similar to that of a previously-published evaluation of the entire cohort [31]. Although sample size precludes direct statistical analysis of prodromal intervals in MSA, substantial differences with Lewy body disease are evident, notably normal olfaction at all intervals, normal cognition, an unclear/highly variable autonomic interval with absent orthostatic hypotension before diagnosis, and a shorter motor interval (e.g., UPDRS Part III interval =≅2 years in MSA on average, versus 6.5 years in the entire cohort).
DISCUSSION
The prospective nature of our cohort enabled us to directly and comprehensively measure the emergence of MSA-P from its prodromal stages. We report here the evolution of 4 prodromal MSA-P patients, tracked 1.3–6 years before phenoconversion to parkinsonism. Although conclusions are obviously limited by the low number of cases, we note several themes, with potential implications for early MSA diagnosis and potential future MSA criteria:
It is important to emphasize that we are observing one pathway of prodromal MSA, namely that which emerges from prodromal RBD. Most observations of early/prodromal MSA come from clinics specializing in autonomic dysfunction [2–4]. These prior studies have been either retrospective or included a follow-up examination that is less comprehensive than the current study, making direct comparison to our findings impossible. Regardless, our findings emphasize the importance of avoiding over-reliance on one pathway to make prodromal diagnosis (whether that be from autonomic clinics or sleep/RBD clinics); in particular, our finding that clinically-evident autonomic function is not an invariable feature of early MSA would have not been possible to observe using autonomic clinic-based studies.
Some limitations should be noted. The primary limitation is a small sample size, which limits statistical analysis. This reflects the relative rarity of MSA, with the inherent difficulty in detecting its prodromal stage. This limitation is counterbalanced by the fact that there is no published prospective study of prodromal MSA that has assessed patients with such a comprehensive testing protocol. Another important limitation is that the diagnosis of MSA cannot be directly confirmed. In many cases, ancillary diagnostic investigations were conducted, but these were at the discretion of the clinical team (once patients phenoconvert there is no formal research follow-up protocol, although we did follow patients whenever possible). Autonomic testing was very limited in our protocol to simple clinical measures; it is possible that numerous abnormalities would have been documented with a detailed laboratory-based autonomic protocol. Finally, we are missing a 3-year period of prodromal data from one patient, who had elected to not follow with us until he had symptoms.
In summary, by systematically observing MSA emerging from prodromal stages of RBD, we have observed a prodromal state characterized by rapid motor progression, without inevitable clinical autonomic dysfunction, and with normal olfaction and cognition. These findings may have implications for detection of early stages of MSA and for development of prodromal MSA criteria.
Footnotes
ACKNOWLEDGMENTS
This study was funded by the Fonds de Recherche du Québec – Santé, the W. Garfield Weston Foundation, and the Canadian Institute of Health Research.
CONFLICT OF INTEREST
Dr. Postuma reports grants and personal fees from Fonds de Recherche du Québec – Santé, the Canadian Institute of Health Research, Parkinson Canada, the W. Garfield Weston Foundation, the Michael J. Fox Foundation, the Webster Foundation, and Roche, and personal fees from Takeda, Roche/Prothena, Teva Neurosciences, Novartis Canada, Biogen, Boehringer Ingelheim, Theranexus, GE HealthCare, Jazz Pharmaceuticals, Abbvie, Jannsen, and Otsuko, outside the submitted work. Dr. Gagnon received grants from the Fonds de Recherche du Québec – Santé, the W. Garfield Weston Foundation, and the Canadian Institutes of Health Research. He holds a Canada Research Chair in Cognitive Decline in Pathological Aging. Dr Montplaisir received personal compensation as consultant (Impax pharma, Servier, Jazz pharma, Merk, Valeant), speaker (Valeant), and received financial support for research activities from Merck, GlaxoSmithKline is funded by grants from the Fonds de Recherche du Québec – Santé, the W. Garfield Weston Foundation, and the Canadian Institutes of Health Research. A Pelletier has nothing to disclose.