
Abstract
Parkinson’s disease is a debilitating neurodegenerative disorder whose etiology is still unclear, hampering the development of effective treatments. There is an urgent need to identify the etiology and provide further effective treatments. Recently, accumulating evidence has indicated that infection may play a role in the etiology of Parkinson’s disease. The infective pathogens may act as a trigger for Parkinson’s disease, the most common of which are hepatitis C virus, influenza virus, and Helicobacter pylori. In addition, gut microbiota is increasingly recognized to influence brain function through the gut-brain axis, showing an important role in the pathogenesis of Parkinson’s disease. Furthermore, a series of anti-infective agents exhibit surprising neuroprotective effects via various mechanisms, such as interfering with α-synuclein aggregation, inhibiting neuroinflammation, attenuating oxidative stress, and preventing from cell death, independent of their antimicrobial effects. The pleiotropic agents affect important events in the pathogenesis of Parkinson’s disease. Moreover, most of them are less toxic, clinically safe and have good blood-brain penetrability, making them hopeful candidates for the treatment of Parkinson’s disease. However, the use of antibiotics and subsequent gut dysbiosis may also play a role in Parkinson’s disease, making the long-term effects of anti-infective drugs worthy of further consideration and exploration. This review summarizes the current evidence for the association between infective pathogens and Parkinson’s disease and subsequently explores the application prospects of anti-infective drugs in Parkinson’s disease treatment, providing novel insights into the pathogenesis and treatment of Parkinson’s disease.
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disease worldwide, ranking only after Alzheimer’s disease [1]. The incidence of PD in people aged 65 years and older reaches 2% and increases rapidly with age [2]. Pathologically, PD is characterized by the loss of nigrostriatal dopamine neurons and the formation of Lewy bodies [3]. However, the etiology and underlying pathogenesis remain unknown. A range of genetic and environmental factors appear to play a role in the pathogenesis of PD [4]. It is generally believed that oxidative stress, α-synuclein (α-syn) aggregation, neuroinflammation, and mitochondrial dysfunction are implicated in the pathogenesis [5–7]. However, to date, there is no cure or disease-modifying agent available for PD. The current drugs for the treatment of PD mainly alleviate motor symptoms, but they cannot slow the progression of the disease [8]. Therefore, it is imperative to determine the actual etiology and develop drugs that can cure the disease at its source.
Accumulating evidence has revealed that infection may play a critical role in neurological diseases, such as herpes simplex virus in Alzheimer’s disease [9], Enterovirus [10] and human herpesvirus [11] in amyotrophic lateral sclerosis, and Zika virus in microcephaly [12]. Since the outbreak of encephalitic lethargica (EL) and post-encephalitic Parkinsonism (PEP) after the 1918 H1N1 influenza pandemic [13, 14], the possibility of an infective etiology for PD has been intensely discussed [15] (Fig. 1). Apart from this, many recent studies have reported that traditional anti-infective drugs exhibit novel neuroprotective effects, which are separate from their antimicrobial effects, showing a promising potential in treating PD [16, 17].
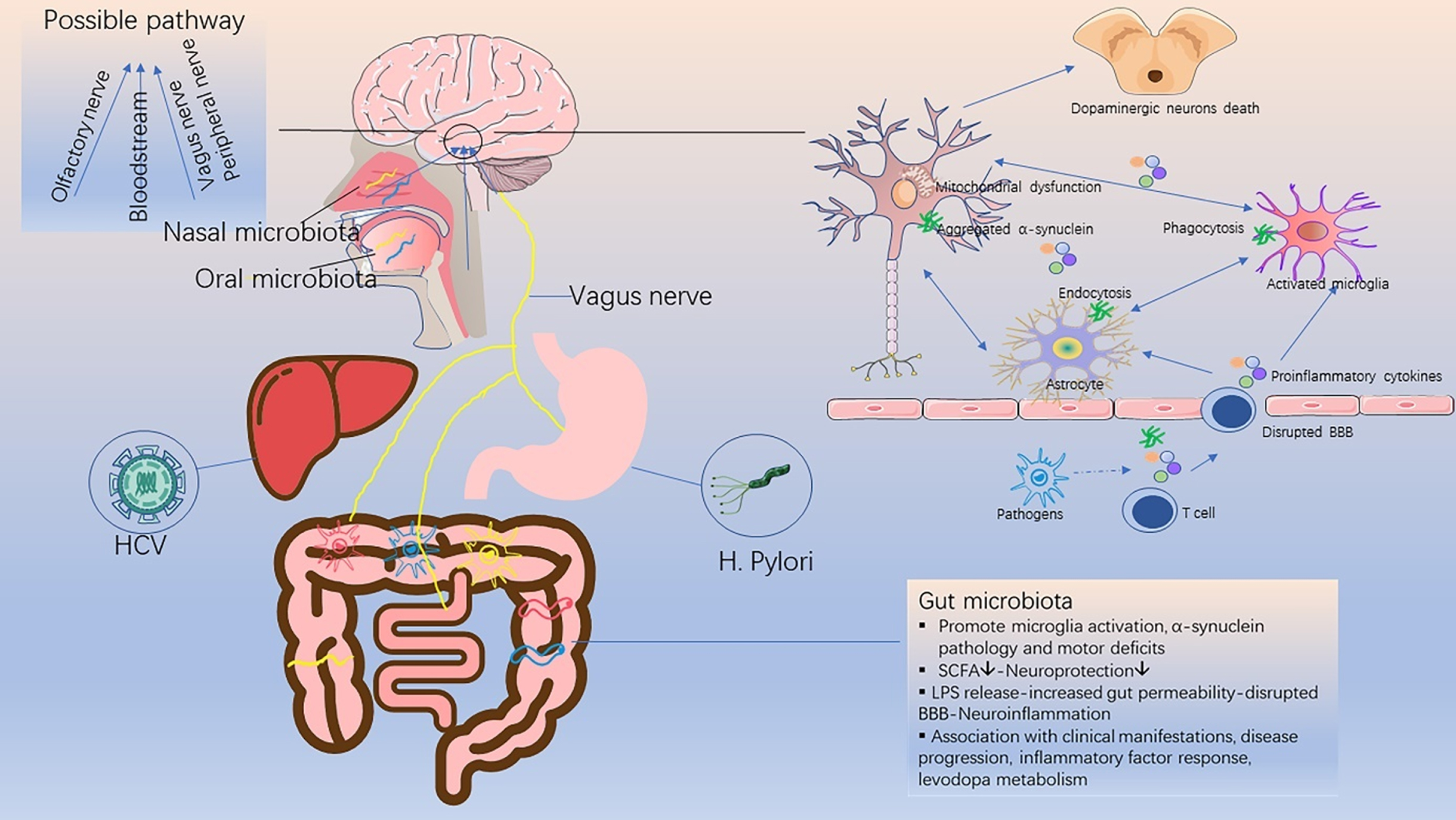
Schematic diagram of the relationship between infective pathogens and Parkinson’s disease. Infective pathogens enter the central nervous system through the bloodstream or nerves, causing inflammatory response and blood-brain barrier disruption through the release of pro-inflammatory cytokines. This subsequently leads to a series of glial activation, neuroinflammation, α-synuclein accumulation and neuronal death, which trigger or accelerate the onset of PD. SCFA, short chain fatty acids; LPS, lipopolysaccharide; BBB, blood-brain barrier.
This review aims to summarize the current evidence for the association between infective pathogens and PD (Table 1), highlight recent studies with anti-infective agents in PD (Table 2), and provide new insights into the pathogenesis and treatment of PD.
Association of infective pathogens in the development of Parkinson’s disease and possible pathogenesis
VZV, varicella zoster virus; JEV, Japanese encephalitis virus; WNV, West Nile virus; BBB, blood-brain barrier; CNS, central nervous system; EL, encephalitic lethargica; PEP, post encephalitic Parkinsonism.
Neuroprotective effects and possible mechanisms of anti-infective agents in Parkinson’s disease
INFECTIVE PATHOGENS ASSOCIATED WITH PARKINSON’S DISEASE
Viral infection and Parkinson’s disease
Hepatitis C virus (HCV)
HCV is an enveloped, single-stranded RNA virus that causes a major public health problem worldwide. In addition to liver injury, chronic HCV infection also causes a series of extrahepatic manifestations, including fatigue, depression, cognitive dysfunction, diabetes, atherosclerosis, and stroke [18, 19].
Recently, emerging data from epidemiological studies has indicated that HCV infection is associated with PD as a risk factor. The HCV-infected patients in most of these studies usually had a significantly increased risk of developing PD [20–25], except a study using the U.S. Medicare database [26]. Furthermore, an extensive meta-analysis involving five studies with 323, 974 participants found a higher risk of PD among HCV-infected patients [27]. Remarkably, interferon-based antiviral therapy has recently been shown to reduce the risk of developing PD in patients with HCV infection [28, 29].
It was demonstrated that essential HCV receptors were expressed on brain microvascular endothelial cells, a major component of the blood-brain barrier (BBB), which could provide a gateway for HCV to invade the central nervous system (CNS) and cause neuroinflammation [30]. This notion was supported by the detection of HCV RNA sequences in postmortem brain tissue from infected patients [31]. In a previous study, it was found that both serotonergic and dopaminergic neurotransmissions were altered in HCV-infected patients with chronic fatigue and cognitive impairment [32]. In addition, it was HCV, not HBV, that was found to induce 60% dopaminergic neuron death in the midbrain neuron-glia coculture system in rats, suggesting that HCV had neurotoxic effects on dopaminergic neurons [20]. Furthermore, HCV infection could induce the release of substantial inflammatory cytokines, indicating that neuroinflammation may play a role in PD pathogenesis [20].
Influenza virus
According to clinical discovery, influenza virus may be related to PD, but this relationship is still controversial. The causal link between influenza virus and PD stems from the outbreak of EL and PEP following the 1918 H1N1 influenza pandemic [13]. It was also reported that there was a positive relationship between severe influenza and PD, as well as an inverse relationship between influenza vaccination and PD (although the latter was not statistically significant) [33]. In a large observational study, influenza infection was shown to have a link with PD-like symptoms, such as tremor or gait disturbances, especially in the first few weeks after the diagnosis or documentation of influenza infection [34]. In addition, the risk of developing parkinsonism symptoms was associated with recent influenza, number of influenza episodes, and severity of preceding influenza infections, which indicated that influenza-related neuronal damage may be a cumulative process [34]. What’s more, a synergistic effect of influenza and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) induced the loss of dopaminergic neurons in mice, which could be eliminated by influenza vaccination or treatment with oseltamivir carboxylate [35]. This further supports the link between influenza virus and PD.
However, the pathology and manifestations of PEP differ from those of idiopathic PD [36], and influenza genes were not detected in the brains of EL patients from 1916 to 1920 [37], which did not support the role of influenza virus as a causative agent of PD.
On the other hand, an array of proinflammatory mediators were elevated in patients with influenza virus-associated encephalopathy, indicating the role of inflammation in the pathogenesis of PEP [38]. An animal study found that the highly pathogenic H5N1 influenza virus could enter the CNS from the peripheral nervous system and activate the innate immune response in the brain, leading to a significant increase in phosphorylation and aggregation of α-synuclein and dopaminergic neuron degeneration [39]. Although the transient loss of tyrosine hydroxylase-positive (TH+) dopaminergic neurons was found to have largely recovered by 90 days post-infection, a long-lasting inflammatory response, especially permanent microglia activation, remained in H5N1-infected mouse models [40]. Moreover, a recent study demonstrated that H1N1 influenza A viral infection and replication could lead to the formation of α-synuclein aggregates [41].
Varicella zoster virus (VZV)
The initial infection of VZV causes varicella, which then lies dormant in the body and causes herpes zoster when reactivated. Early studies have suggested that there was no link between herpes zoster and PD. However, small sample sizes and bias due to self-reported infection history may limit the results. Recently, two cohort studies found a positive correlation between herpes zoster and the risk of PD, whether in elderly or midlife patients [42, 43]. Neuroinflammation and immunological changes involved in both conditions may explain the association. On the other hand, chickenpox infection was reported to have no association with PD [44]. However, in one study, chickenpox infection in childhood was found to be negatively correlated with PD, showing a possible protective effect [33]. The reverse relationship between childhood chickenpox infection and PD is difficult to explain, likely because the immune response activated at a younger age is important in reducing the possibility of viral infection that may damage dopaminergic neurons.
Japanese encephalitis virus (JEV)
Encephalitis caused by JEV is endemic throughout Asia each year, which could cause a high frequency of movement disorders. Japanese encephalitis (JE) can lead to a transient form of parkinsonism, which is characterized by varying severity of rigidity, hypokinesia, masking face, prominent hypophonia and a lower frequency of tremor [45]. Neuronal loss with gliosis in the substantia nigra pars compacta was observed in Fischer rats infected with JEV, which was similar to the pathological changes in PD [46]. Additionally, these rats showed noticeable bradykinesia and improved behavior after L-dopa administration. In addition, decreased dopamine levels and neuropathological characteristics with aging were also observed in JEV-induced PD rat models [47]. A significant decrease in the concentrations of norepinephrine, dopamine, and homovanillic acid in the cerebrospinal fluid was also noticed in JE patients [48], which may be caused by severe structural damage to the thalamus, basal ganglia, and brainstem, as detected on magnetic resonance imaging (MRI).
West Nile virus (WNV)
Movement disorders, especially tremor, myoclonus, and parkinsonism, may be unrecognized manifestations of acute WNV disease [49]. Parkinsonian features associated with acute WNV disease were transient in most cases and diminished over time, with a generally good prognosis [50]. On rare occasions, WNV infection can spread to the CNS in humans, which mechanisms are not yet completely understood. WNV encephalitis could result in immune responses in the brain and neuronal apoptosis. MRI showed abnormalities in the bilateral basal ganglia, thalamus, and pons in patients with severe persistent parkinsonism symptoms [51]. In addition, an increased expression of α-syn was observed in WNV-infected primary neurons. α-syn knockout mice also showed increased viral titers, increased neuronal damage, and accelerated mortality following the introduction of WNV [52]. Moreover, it was demonstrated that α-synuclein expression in intestinal neurons increased with gastrointestinal infection and induced leukocyte migration and dendritic cell maturation, suggesting the role of α-synuclein in gastrointestinal immunity [53]. The aforementioned aspects unveil the novel and significant role of α-synuclein in suppressing viral infection. It also further indicates that the acute onset of parkinsonism during WNV encephalitis may be due to WNV-induced dopaminergic neuronal death.
Western equine encephalitis virus (WEEV)
WEEV is a mosquito-borne arbovirus that can cause fatal encephalitis in humans and horses. Early studies have reported that WEEV infection could cause parkinsonian sequelae following encephalitis in humans [54]. The intranasal inoculation of recombinant WEEV in outbred CD-1 mice could cause significant neural invasion, resulting in the dissemination of the virus from the olfactory bulb to the basal midbrain [55]. The non-lethal encephalitic infection with WEEV led to sustained loss of dopaminergic neurons, persistent glial cell activation, formation of proteinase k-resistant α-synuclein aggregates, and gene expression profiles consistent with a neurodegenerative phenotype in mice. A subsequent study built on this result found that astrocytes played a key role in the initiation of PD-like pathology via the innate immune inflammatory pathway following WEEV infection [56].
Human immunodeficiency virus (HIV)
HIV, also known as acquired immunodeficiency syndrome (AIDS) virus, mainly attacks CD4 + T lymphocytes and macrophages in the human body. People infected with HIV often develop CNS disorders, commonly referred to as AIDS dementia complex (ADC). Psychomotor slowing, apathy, and motor disorders, such as bradykinesia, postural instability, gait abnormalities, and hypomimetic facies, were common manifestations of ADC [57]. It was found that HIV was preferentially present in inflammatory infiltrates and glial cells of the basal ganglia, including the substantia nigra [58]. Additionally, both dopamine and its major metabolite homovanillic acid were significantly reduced in the AIDS group [59]. This finding was consistent with the loss of dopaminergic neurons in the substantia nigra, which may be the basis of motor dysfunction in AIDS patients [60, 61]. Besides, an autopsy study found that the frequency of α-synuclein in the brains of HIV-infected patients was significantly higher than that of the healthy group, indicating that HIV patients may be more prone to develop PD [62]. In addition, HIV had an effect on PD-related proteins, such as DJ1 [63] and LRRK2 [64]. Moreover, a decrease in AIDS-related parkinsonism was observed after highly active antiretroviral therapy [65].
SARS-CoV-2
A serious threat to global health has been posed with the emergence of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, coronavirus disease (COVID-19). Although respiratory symptoms are a prominent feature of COVID-19, SARS-CoV-2 can also invade other organs and affect multiple systems in the body. According to recent studies, SARS-CoV-2 could invade and attack the nervous system [66], causing neurological symptoms [67] and neuropathological damage [68]. Anosmia is a common early symptom of COVID-19 [69], and is also considered an early sign of PD. It is still unknown if this reflects an effect of SARS-CoV-2 on the peripheral nervous system, such as the olfactory nerve, or SARS-CoV-2 could further infect the CNS through the nasal cavity, a potential pathway into the CNS [70].
Compared with the 1918 influenza pandemic and avian influenza, reports of SARS-CoV-2-related encephalopathy have been slow to emerge. Until now, there have been relatively few reports of parkinsonism following SARS-CoV-2 infection. A 45-year-old man [71] and a 35-year-old woman [72] developed new asymmetric parkinsonism symptoms after SARS-CoV-2 infection. Both patients also showed radiographic abnormalities of dopamine dysfunction in the presynaptic striatum and responded well to levodopa. Interestingly, a 58-year-old man developed acute hypokinetic-rigid syndrome after SARS-CoV-2 infection and showed spontaneous improvement after 14 days [73]. According to an Italian community-based PD cohort study, motor and non-motor symptoms of the COVID-19 group significantly worsened [74]. However, unlike WNV, SARS-CoV-2 infection did not cause upregulation of serum and cerebrospinal fluid α-synuclein levels, regardless of the presence or absence of neurological symptoms in these COVID-19 patients [75].
Considering the far-reaching impact of SARS-CoV-2, further longitudinal studies are urgently needed.
Bacterial infection and Parkinson’s disease
Helicobacter pylori (H. pylori)
H. pylori is a helically-shaped, microaerophilic, gram-negative bacterium affecting approximately half of the world’s population. It is well-known that gastrointestinal symptoms precede motor symptoms in PD patients [76]. Besides, a follow-up study demonstrated that PD patients had a higher incidence of ulcers, which preceded parkinsonian symptoms by approximately 10 years [77]. Therefore, the relationship between H. pylori infection and PD has recently been extensively explored [78].
Increasing evidence demonstrated that PD patients had a higher prevalence of H. pylori infection. In one study, PD patients had a three-fold elevated risk of H. pylori seropositivity, compared with the control group [79]. Interestingly, their siblings were also more likely to suffer from parkinsonism symptoms and H. pylori positivity. An extensive meta-analysis involving 33, 125 participants revealed that a 1.5–2 fold increased risk of developing PD in H. pylori-infected persons [80]. Besides, it has been repeatedly reported that there existed a positive correlation between H. pylori infection and worse motor function in PD patients [81]. What’s more, eradication of H. pylori could ameliorate motor symptoms in PD patients, which was recommended in PD patients treated with levodopa because it may improve drug bioavailability and reduce motor fluctuations [82]. With the clearance of H. pylori, the levodopa onset time, ON duration, motor severity, and life quality all got improved [83]. A subsequent comprehensive meta-analysis found significant associations between H. pylori infection and higher Unified Parkinson’s Disease Rating Scale (UPDRS) scores, as well as a significantly lower UPDRS score in H. pylori-eradicated PD patients [84].
Overall, these results further confirm the view that H. pylori infection may play a key role in the course or even in the pathogenesis of PD [78]. However, the exact mechanism remains unclear. It is generally believed that possible pathogenic mechanisms include toxins produced by H. pylori, the disrupted gut microbiota [85], substantial proinflammatory cytokines [86, 87], the molecular mimicry between H. pylori and proteins essential for normal neurological functions [88, 89], in addition to the pharmacokinetic effects of H. pylori on levodopa [90–92].
Microbiota and Parkinson’s disease
Gut microbiota
Gut microbiota is composed of complex microbial communities. Recently, it has been increasingly recognized that gut microbiota affects brain function through the gut-brain axis. Emerging evidence suggests that there is a link between gut microbiota and PD. Gut microbiota was necessary for motor deficits, microglial activation, and α-synuclein pathology using α-synuclein overexpression mouse model [93]. Orally taken specific microbial metabolites promoted neuroinflammation and motor dysfunction in germ-free mice. The transplantation of gut microbiota from PD patients into germ-free α-syn overexpression mouse models exacerbated the motor deficits in mice, while antibiotic treatment reversed these effects, suggesting the critical role of gut microbiota in the pathogenesis of PD.
A number of studies have reported differences in the composition of the gut microbiota between PD patients and healthy controls [94]. It has also been shown that not only the concentration of beneficial fecal short chain fatty acids (SCFA), but also the abundance of SCFA-producing bacteria were lower in the PD group than in the control group [95, 96]. Bacteria that produce SCFA, particularly butyrate, may be an important factor in the pathogenesis and progression of PD. A link was found between PD clinical characteristics and the gut microbiota [97]. In this study, the genera Escherichia/Shigella were negatively associated with disease duration. The genera Butyricicoccus and Clostridium XlVb were associated with cognitive impairment. The genera Dorea and Phascolarcto bacterium were negatively associated with levodopa equivalent doses. In a two-year follow-up study, a more common Firmicutes-dominant enterotype and a lower abundance of Prevotella genus were observed in faster-progressing PD patients, compared with slower-progressing patients and controls, indicating a connection between gut microbiota and disease progression [98]. Altered gut microbiota was also found to be associated with the plasma cytokine response, in addition to clinical phenotype and severity [99]. Intriguingly, gut microbiota not only affects the health status of the host, but also the effectiveness of drug treatment [100, 101].
Oral microbiota
Comparing the oral microbiota of 72 PD patients and 76 healthy controls using 16S rRNA gene amplicon sequencing, significant differences were observed in the beta diversity and abundance of individual bacterial taxa [102]. Additionally, it was found that patients with periodontal inflammatory disease (PID) had a significantly higher risk of developing PD [103], while dental scaling over five consecutive years could reduce the risk of developing PD in individuals without PID, which emphasized the value of early and consecutive dental scaling in preventing the development of PD [104]. Moreover, PD patients tended to have poorer oral health, lower frequency of daily tooth-brushing, longer gaps between dental visits, and reduced salivary flow [105]. Even if confounding factors were controlled, PD patients still had more missing teeth, caries, dental plaque, cariogenic bacteria in saliva, and poorer periodontal health [106]. Individuals with poor oral health also tended to have higher Hoehn and Yahr scores and lower MMSE scores [107].
Nasal microbiota
The olfactory bulb was affected by the pathology of α-synuclein in the early stage, presenting as hyposmia. As assumed in the “dual-hit” hypothesis, the nasal cavity acts as an important portal for pathogens to spread to the CNS, which may be involved in the pathogenesis of PD [108].
After comparing the nasal microbiota using the 16S rRNA gene amplicon sequencing approach, no significant difference in alpha or beta diversity was observed between 69 PD patients and 67 healthy controls in one study [102]. Similar results were found in another study [109]. However, owing to the limited number of studies and patients, we cannot provide a definitive answer on whether the nasal microbiota is related to PD.
ANTI-INFECTIVE AGENTS AND PARKINSON’S DISEASE
Antiviral agents and Parkinson’s disease
Interferon
Wangensteen et al. identified that two patients developed PD in the short term during treatment of hepatitis C with interferon alpha [110]. Intriguingly, two recent epidemiological studies have shown that interferon treatment could reduce the risk of PD in patients with HCV infection [28, 29]. One study demonstrated that the reduced PD risk appeared to be obvious at five years after antiviral therapy and became more statistically significant at the end of follow-up [28]. Another study found that after treatment with PegIFN/RBV antiviral therapy, the risk of developing parkinsonism decreased by 38% [29]. One possible explanation for this discrepancy is that the two patients with hepatitis C in the case reports were already in the premotor stage, and antiviral treatment could not prevent their eventual progression to PD.
Amantadine
From 1966 to the present, amantadine experienced a transition from an anti-flu drug to an anti-parkinsonian agent [111]. In 1969, it was accidentally discovered that a female patient with PD improved motor symptoms after taking amantadine for flu, while her symptoms worsened after stopping the medication [112, 113]. Amantadine seemed to be most effective in the early stages of PD and had the greatest impact on tremors [114]. The effect of amantadine on dyskinesia has also been confirmed in multiple studies [115]. The mechanism of amantadine in treating PD may involve enhancing dopamine release from presynaptic terminals [116] and blocking N-methyl-D-aspartate (NMDA) receptors [117]. In addition, its mild anticholinergic effect has been reported [118].
ADS-5102, the amantadine extended-release capsule, has been demonstrated in several pivotal trials as an effective treatment for reducing OFF-time and dyskinesia [119]. Thus, it has recently been approved for the treatment of dyskinesia in PD patients by FDA [120].
Antimicrobial agents and Parkinson’s disease
Ceftriaxone
Ceftriaxone, a third-generation cephalosporin in the β-lactam antibiotic group, is a well-tested and safe drug that has been used for the treatment of a number of bacterial infections for several decades. Recently, the neuroprotective effects of ceftriaxone were observed in a wide range of neurological disorders, including Alzheimer’s disease, PD, amyotrophic lateral sclerosis, Huntington’s disease, cerebral ischemia, seizure, pain, and spinal cord injury [121].
It was proved that ceftriaxone could reduce L-dopa-induced dyskinesia severity in a 6-hydroxydopamine (6-OHDA) PD model [122]. In MPTP rat models, post-treatment with ceftriaxone significantly improved motor deficits and attenuated oxidative damage, as well as pro-inflammatory cytokines [123]. Additionally, ceftriaxone was shown to significantly restore the decreased activity of brain-derived neurotrophic factor (BDNF) in the striatum of MPTP-treated rats [124]. In a 6-OHDA PD rat model, ceftriaxone was reported to increase glutamate uptake, upregulate GLT-1 expression and reduce striatal tyrosine hydroxylase loss [125]. And it has been shown that ceftriaxone protected astrocytes from 1-methyl-4-phenylpyridinium (MPP+)-induced neurotoxicity by suppressing the NF-κB/JNK/c-Jun pathway [126]. In an MPTP-induced rat model of PD dementia, ceftriaxone not only reversed behavioral deficits but also prevented the loss of neurogenesis [127]. Another study concluded that ceftriaxone had an effect on behavioral and neuronal changes, which could prevent hippocampal cell loss and improve cognitive function in a PD rat model [128]. Furthermore, ceftriaxone could bind to α-synuclein with good affinity and block its polymerization in vitro.
Doxycycline
As a member of the tetracycline antibiotic family, doxycycline is used to treat a variety of bacterial infections, such as acne and intestinal infections. In clinical practice, it is widely used for its protective property with clinical safety records, increased lipid solubility, and excellent penetration of the BBB.
It has been demonstrated that doxycycline exhibits neuroprotective effects on PD both in vivo and in vitro, with MPTP and 6-OHDA animal models. In a 6-OHDA PD model, doxycycline blocked 6-OHDA neurotoxicity and conferred neuroprotection by inhibiting glial activation [130], the mechanism of which may be mediated by suppressing the p38 MAPK and NF-κB signaling pathways [131]. Doxycycline was neuroprotective against nigral dopaminergic degeneration by a dual mechanism, anti-apoptotic and anti-inflammatory mechanisms involving the downregulation of matrix metalloproteinase-3 (MMP-3) [132]. In addition, doxycycline not only inhibited the fibril formation of amyloidogenic proteins such as Aβ peptide, PrP peptide, and β-microglobulin, but also affected the aggregation of α-synuclein and the seeding of new oligomers. It was determined that doxycycline reshaped α-synuclein oligomers into off-pathway species, with parallel β-sheet content and a less hydrophobic surface, which did not evolve into fibril formation [133]. Furthermore, it is suggested that doxycycline at sub-antibiotic doses would be sufficient to exert anti-inflammatory activity and neuroprotective effects without changing bacterial susceptibility to antibiotics [134].
Minocycline
Minocycline, a semisynthetic second-generation tetracycline, exerts anti-inflammatory properties that are independent of its antimicrobial effects. Minocycline was shown to prevent nigrostriatal dopaminergic neurodegeneration. In the MPTP mouse model of PD, minocycline treatment blocked the MPTP-induced loss of striatal dopamine and its metabolites by inhibiting MPTP-induced glial iNOS, caspase 1 expression in vivo and NO-induced neurotoxicity in vitro [135]. In another study, minocycline inhibited MPTP-induced microglia activation and prevented the production of microglial-derived deleterious mediators, thereby reducing the loss of nigrostriatal dopaminergic neurons and the formation of nitrotyrosine produced by MPTP [136]. The behavioral and morphological effects of minocycline on PD have also been reported in the 6-OHDA rat model. Rats receiving minocycline displayed lower rotations, reduced tyrosine hydroxylase-positive cell loss, and increased nigral cell size and fiber density [137]. Minocycline conferred neuroprotection against NMDA neurotoxicity by inhibiting microglia. And the neuroprotective effect was associated with the inhibition of p38 MAPK in microglial cells [138].
However, beyond these promising results, the deleterious effects of minocycline on PD have also been reported [139–141]. Minocycline significantly exacerbated MPTP-induced damage to dopaminergic neurons, although it inhibited microglial activation [139]. This differs from previous studies in which minocycline inhibited microglial activation to exert neuroprotective effects [136]. The discrepancy may be attributed to the fact that minocycline may have variable or even deleterious effects depending on the route of administration, drug concentration or dosing interval. In addition, inhibiting microglia is not always beneficial since this may compromise its protective and reparative functions at the same time.
Rifampicin
Rifampicin, a semi-synthetic derivative of rifamycins, is widely used for the treatment of tuberculosis and leprosy. Rifampicin was able to prevent neuronal degeneration and significantly increase the surviving dopaminergic neuron numbers after MPP+intoxication in vitro [142]. Moreover, it was proved to attenuate the MPTP-induced neurotoxicity in the mouse brain [143]. Rifampicin has also been shown to inhibit α-synuclein fibrillation and disaggregate existing fibrils in a concentration-dependent manner [144]. This is consistent with a subsequent study in which rifampicin pretreatment caused a concentration-dependent increase in cell viability and a reduction in α-synuclein expression [145]. In addition, rifampicin-induced neuroprotection was suggested to be associated with its free radical scavenging activity [146]. It was found that rifampicin pretreatment could protect PC12 cells against cell death by suppressing rotenone-induced apoptosis and ameliorating mitochondrial oxidative stress [147]. It was also found that rifampicin could significantly inhibit the production of pro-inflammatory mediators in lipopolysaccharide-stimulated BV2 cells [148]. Moreover, the anti-inflammatory effects of rifampicin were mediated by the inhibition of NF-κB via the regulation of the IκB pathways as well as the suppression of MAPKs phosphorylation. Additionally, a recent study using human microglia cells demonstrated that rifampicin attenuated rotenone-induced microglial inflammation by improving autophagy flux and lysosomal function [149, 150].
Furthermore, rifampicin quinone, the oxidized form of rifampicin, is proved more potent than the parent compound itself, especially in reducing microglial inflammatory responses [151]. And the anti-inflammatory effects of these two drugs were related to the inhibition of PI3K-dependent and independent signaling events.
Geldanamycin (GA)
GA, a benzoquinone antibiotic isolated from the bacterium Streptomyces hygroscopicus, specifically binds to and inhibits the function of the molecular chaperone heat shock protein 90 (Hsp90), which promotes proteasomal degradation. GA has been shown to protect against α-synuclein-induced dopaminergic neuronal loss in the Drosophila model of PD [152]. Interestingly, previous studies have also shown that the molecular chaperones could suppress protein misfolding and aggregation, thus exerting protective effects against neurodegenerative diseases, including Huntington’s disease and spinal-cerebellar ataxia [153, 154]. It has been demonstrated that GA could induce Hsp70, prevent α-synuclein aggregation, and protect against α-synuclein-induced toxicity in vitro [155]. Although GA could selectively inhibit Hsp90, upregulate Hsp70, and reduce α-synuclein-induced neurotoxicity, its application is limited due to its hepatotoxicity and poor brain permeability. However, its structural analogs, such as 17-(allylamino)-17-demethoxygeldanamycin (17-AAG) and 17-dimethylaminoethylamino-17-demethoxy-geldanamycin (17-DMAG) exhibited greater potency, reduced toxicity, and improved brain permeability. Additionally, SNX-9114, a novel Hsp90 inhibitor, was reported to significantly rescue dopamine levels in the striatum after chronic treatment in a rat model of parkinsonism, although it failed to protect against α-synuclein-induced nigrostriatal toxicity [156].
Anti-fungal agents and Parkinson’s disease
Rapamycin
As an inhibitor of mTOR, rapamycin can effectively initiate the autophagy process and remove the accumulated denaturing proteins and damaged organelles of cells, which play a crucial role in the pathogenesis of neurodegenerative diseases.
Rapamycin has been verified to confer neuroprotection against PD toxins both in vitro and in vivo [157]. It could protect against neuronal death by blocking mTORC1–dependent translation of the pro-cell death protein RTP801. Rapamycin was reported to reduce α-synuclein accumulation and promote oligomer clearance via enhanced autophagy [158]. In addition, pretreatment with rapamycin could improve animal behaviors and decrease dopaminergic neuron loss in the 6-OHDA-induced rat model, which might be due to the property of rapamycin on protecting mitochondria from oxidative stress and apoptosis [159]. The effects of rapamycin on glia and anti-inflammation have been further verified in a study [160], wherein the astrocytic upregulation of glutamate transporters and improvement in IL-6 expression were observed in the MPTP models. Furthermore, the activation of the mTOR-Akt-NF-κB cascade, in part the JAK2/STAT3 pathway, appeared to be crucial in mediating the biological effects of rapamycin.
These findings suggest that rapamycin could be an effective and promising neuroprotective candidate for the treatment of PD (Fig. 2). However, the adverse effects of rapamycin as an immunosuppressive agent for long-term treatment should not be ignored. Long-term use of rapamycin could lead to lung toxicity [161] and increase the risk of type 2 diabetes [162].
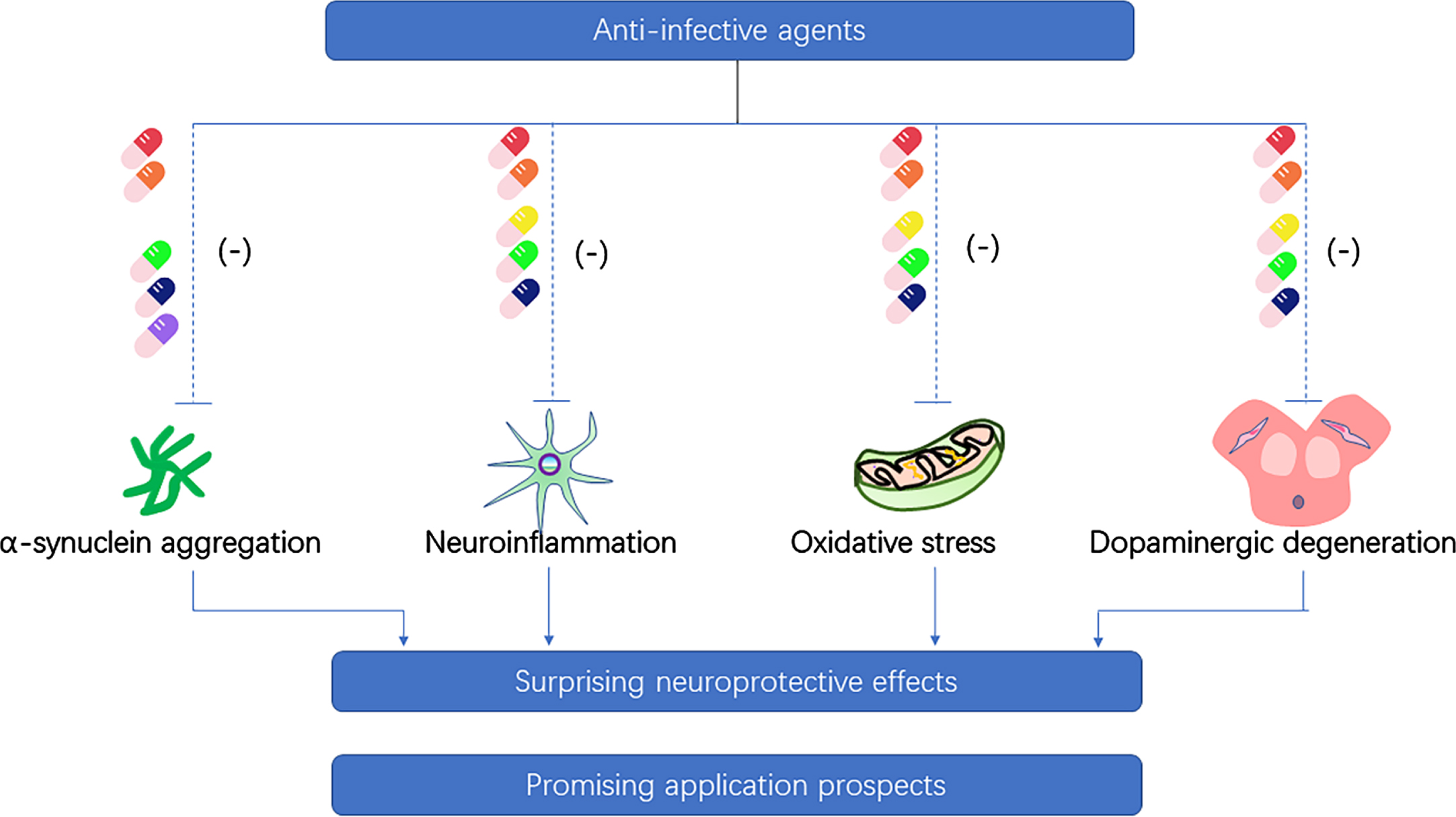
Neuroprotective property of anti-infective agents on the pathogenesis of Parkinson’s disease. (Ceftriaxone-red pill, doxycycline-orange pill, minocycline-yellow pill, rifampicin-green pill, rapamycin-dark blue pill, geldanamycin-purple pill).
Anti-parasitic agents and Parkinson’s disease
Niclosamide
A recent study has shown that the anthelminthic drug, niclosamide, and its analog, AM85 can activate PINK1 in cells through the reversible impairment of mitochondrial membrane potential, which are expected to be a strategy for the treatment of PD [163]. In addition, niclosamide has been used safely to treat parasitic infections for a long time with no obvious serious adverse reactions.
CONCLUSIONS AND FUTURE PERSPECTIVES
In this review, we present the current evidence for the possibility of infective pathogens in the etiology of PD and the potential use of antibiotics as neuroprotective agents in PD. In the past few years, people’s attention on PD has mainly focused on α-synuclein. However, the etiology of PD remains elusive, hampering the development of effective treatments. Obviously, it is of great importance to identify the etiology and provide further corresponding treatments. Infection provides a fascinating hypothesis for the etiology of PD. As early as the 1920s, PD was suspected to be an infectious disease because it often appeared after EL [13]. In a recent study, it was discovered that the bacterial endotoxin lipopolysaccharide could transform asymptomatic Pink1–/–mice into a fully penetrant PD model, suggesting that intestinal infection may act as a triggering event in PD [164]. These studies highlight that chronic infection with bacterial and viral pathogens may be a trigger for PD. As an initiating factor, infection may cause a range of gut microbiota dysbiosis, glial activation, neuroinflammation, and α-synuclein accumulation, which may elicit or accelerate the pathogenesis of PD and lead to its occurrence. The underlying cause for PD with infection is unknown. Controversy still exists as to the potential pathogenesis of infection with PD. In addition, whether PD patients become ill as a result of infecting pathogens or simply because they are more susceptible to infective factors requires further research.
Currently, there are no cure or disease-modifying drugs that can stop or slow the progression of PD. However, a growing body of research suggests that anti-infective agents appear to be promising candidates for the treatment of PD. These agents affect crucial events in the pathogenesis of PD, such as interfering with α-synuclein aggregation, inhibiting neuroinflammation, attenuating oxidative stress, and preventing from cell death, thereby effectively hindering disease progression at multiple levels and showing promising application prospects. Although the potential for antibiotics in the treatment of PD is encouraging, few clinical trials have been conducted so far. As is known to all, several important idiopathic diseases have been found to be associated with previously unsuspected infections, such as H. pylori in gastric ulcer and gastric cancer [165], human papillomavirus in cervical cancer [166], Epstein-Barr virus in nasopharyngeal cancer [167], and hepatitis B virus in liver cancer [168]. These discoveries have led to new and effective treatments for the disease. If the role of infection in PD can be confirmed, it will provide a new direction and target for the treatment of PD. Besides, drug repurposing can quickly introduce drugs with well-documented safety to new patient populations, significantly speeding up the drug development process [169]. Recently, a nationwide case-control study was carried out, suggesting that early overexposure to antibiotics, particularly antianaerobics and broad-spectrum antibiotics, is associated with subsequent PD, with a delay of 10 to 15 years, which is consistent with the proposed duration of a prodromal period [170]. And this correlation is largely due to alteration in gut microbiota [171, 172]. Further research is needed to clarify the relationship between anti-infective agents and PD. In addition, if further research supports the infective etiology hypothesis for PD, a balance between efficacy and adverse effects should be taken into account. The choice of antibiotics, when and how to use them, the possible antibiotic resistance problem, and the potential impact of antimicrobial prescription on gut microbiota also need further consideration.
Taken together, results from recent studies suggest that infection may play a novel role in the pathogenesis of PD and highlight anti-infective agents as potential therapeutic targets for the treatment of PD. Further epidemiological and experimental studies are needed to confirm this hypothesis.
Footnotes
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China to Dr. Chang-he Shi [grant number 81974211, 81771290, 82171247]; the National Key R&D Program of China to Dr. Yu-ming Xu [2017YFA0105000]; the National Natural Science Foundation of China to Dr. Yu-ming Xu [grant numbers U1904207, 91849115] and the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences [2020-PT310-01].
CONFLICT OF INTEREST
The authors have no conflict of interest to report.