
Abstract
Background:
Lewy body dementia (LBD) has two main phenotypes: Parkinson’s disease dementia (PDD) and dementia with Lewy bodies (DLB), separated by the ‘one-year-rule’. They also show different symptom profiles: core DLB features include fluctuating cognition, REM-sleep behaviur disorder, and visual hallucinations. These symptoms are sometimes present in PDD, representing an intermediate ‘PDD-DLB’ phenotype.
Objective:
DLB-like features may reflect deficits in the functions of the noradrenergic nucleus locus coeruleus (LC). Therefore, we compared the LC in the LBD phenotypes, PD, and controls.
Methods:
38 PD, 56 PDD, 22 DLB, and 11 age-matched control cases from the Parkinson’s UK tissue bank were included. LC tissue sections were immunostained for tyrosine-hydroxylase (TH), α-synuclein, tau, and amyloid-β. TH-neurons were quantified and pathologic burden calculated by %-coverage method.
Results:
The LC shows a stepwise reduction in neuron count from controls, PD, PDD, to DLB. PDD-DLB cases showed an intermediate clinical phenotype that was reflected pathologically. Cell counts were significantly reduced in DLB compared to PDD after correction for demographic factors. LC degeneration contributed significantly to the onset of all DLB symptoms. While α-synuclein was not significantly different between PDD and DLB cases, DLB exhibited significantly less tau pathology.
Conclusion:
DLB and DLB-like symptoms represent noradrenergic deficits resulting from neuronal loss in the LC. PDD and DLB are likely to represent a clinical continuum based on the presence or absence of DLB-like symptoms mirrored by a pathological continuum in the LC.
Keywords
INTRODUCTION
Parkinson’s disease (PD) is a progressive neurodegenerative disorder associated with specific phenotypes of dementia [1]. The dementia syndrome in PD—Lewy body dementia (LBD)—is characterized by visuo-spatial, executive, attentional, and occasional memory deficits [1, 2]. LBD patients may also experience cognitive fluctuations, hallucinosis, and sleep disorders [3, 4]. LBD comprises two main phenotypes of dementia, separated by an arbitrary ‘one-year-rule’. Dementia with Lewy bodies (DLB) is defined by dementia preceding or occurring within one year of the onset of parkinsonism (bradykinesia, tremor, rigidity) [5], whereas PD-Dementia (PDD) is dementia occurring over a year after the onset of parkinsonism [1].
Though some authors argue that DLB and PDD are similar conditions [2], their diagnostic criteria differ in critical ways [1, 5]. The core diagnostic criteria for DLB are fluctuating cognition (FC), visual hallucinations (VH), and REM-sleep Behavioral Disorder (RBD), whereas PDD criteria specify these as ‘associated’ features [1, 5]. Core features for PDD diagnosis are dementia, developing within the context of parkinsonism, with impairment in more than one cognitive domain [1]. The discrepancy in these criteria appears to be well-founded: FC is reported in up to 80%of DLB patients [6] compared to 29%of PDD [7]; RBD is a confirmed risk factor for DLB [5], though there are conflicting studies as to whether this is the case for cognitive decline in PD as well [1, 8–10]; visual hallucinations, though common in PDD, are more prevalent in DLB [11–13]. However, recent studies shown that some PDD cases can mimic DLB. Varanese and colleagues separated PDD patients by the presence of FC and found that fluctuating PDD showed a more similar pattern of cognitive dysfunction to DLB than non-fluctuating PDD [14]. PDD patients without FC showed milder attention and memory deficits, and lower prevalence of hallucinosis.
LBD is characterized by the accumulation of inclusions of aberrant α-synuclein in Lewy bodies and neurites. Whilst LBD is a synucleinopathy, commonly, increased levels of tau and amyloid-β deposition are found in cases of LBD compared to controls and non-impaired PD [15, 16]. Thus, LBD probably reflects a confluence of pathologies. LBD has been shown in previous studies to reflect a later α-synuclein pathologic stage compared to PD without dementia [17, 18], suggesting an increased level of cortical pathology contributes to LBD pathogenesis. However, the precise neuropathological substrates of LBD and LBD symptomatology have remained controversial [19].
The LC is the major source of noradrenaline in the central nervous system, innervating cortical, subcortical and limbic structures [20–22]. In doing so, the LC functions to maintain sleep-wake-cycles, concentration, alertness and attention, thereby promoting the steady cognitive state [23]. Ponto-geniculo-occipital (PGO) waves are electroencephalogram correlates of hallucinosis but are also involved in the initiation of REM-sleep [24]. They originate from the coeruleus/subcoeruleus complex, thereby implicating these regions in DLB [25]. DLB features therefore appear to show deficits in the functions of the LC. In the present study, we will compare the pathology and integrity of the LC neuronal population in DLB and PDD cases, with and without DLB-like features. We will also assess the contribution of the LC to the symptomatology of DLB.
METHODS
Cohort selection and clinical assessment of cases
Clinical data of cases from the Parkinson’s UK Tissue Bank at Imperial College London were compiled by specialist movement disorder neurologists (including R.K.B.P.). Only subjects evaluated by a clinician within two years prior to death and with complete clinical histories were used for this study. The clinical diagnoses of PD, PDD, and DLB were assigned based on published clinical criteria [1, 5]. Only cases with detailed clinical information were used. Retrospective case note evaluation was used to examine cases for pertinent clinical characteristics. This type of analysis is a well-accepted method of case ascertainment and has often been used in clinicopathological studies involving both dementia and parkinsonism [16, 26–28]. While it was not sought to construct post hoc formal diagnoses, all cases were analyzed for evidence of FC, VH, and RBD—the key features of DLB, alongside dementia and spontaneous parkinsonism—thereby creating the three dementia groups in a manner similar to Varanese and colleagues in their study: PDD without DLB features (PDD), PDD with DLB features (PDD-DLB), and DLB [14]. Cases qualified as RBD-positive if there was a formal diagnosis, or high index of suspicion from a specialist in clinical records without formal diagnosis. Additionally, the presence of depression and anxiety disorders were also noted.
Neuropathological assessment
All human tissue work was carried out under the ethical approval held by the Parkinson’s UK Tissue Bank at Imperial College London (REC Ref: 07/MRE09/72). Neuropathological diagnosis was based on α-synuclein, tau and amyloid-β immunohistochemistry. PD cases were confirmed neuropathologically using an established protocol for the neuropathologic diagnosis of Lewy body disease [29]. Cases were excluded based on comorbid clinicopathological diagnosis of AD, and atypical causes of parkinsonism such as multiple system atrophy, progressive supranuclear palsy, and corticobasal degeneration. Control cases were selected based on a lack of clinical neurological diagnoses and a relative lack of neuropathological findings on microscopic examination, including Braak tau stage of III or less [30]. In total, 38 PD, 56 PDD, 22 DLB, and 11 age-matched control cases were included for this study.
Postmortem human brain tissue
Human brain tissues containing the LC were routinely sampled at the rostral pontine level as part of the diagnostic work-up of cases at the Parkinson’s UK Tissue Bank. Samples were fixed for 4 weeks in 10%buffered formaldehyde and then dehydrated in 70%ethanol, 90%ethanol, 100%ethanol and xylene before being embedded in paraffin wax. 7μm sections were cut by microtome, mounted onto microscope slides and dried for 24 h in a 37°C oven in preparation for immunohistochemistry.
Immunohistochemistry
Tissue sections containing the LC were immunostained with anti-Tyrosine Hydroxylase (TH; AB152, EMD Millipore; 1:2000), anti-α-synuclein (BD Transduction 1:300, anti-tau (AT8, ThermoFisher Scientific; 1:1200), and anti-amyloid-β (4G8, Biolegend, 1:15000). In general, slides were dewaxed in xylene twice for 10 min and 5 min, rehydrated in descending concentrations of ethanol (100%, 90%, 70%) for 5 min each before being rinsed in distilled H2O (dH2O). Endogenous peroxidase quenching was carried out using a bath of 1%H2O2 in phosphate-buffered saline (PBS; pH 7.40) for 30 min and slides were rinsed in dH2O again for 5 min. TH and α-synuclein slides pre-treated using heat-induced epitope retrieval in a sodium citrate buffer (pH 6), amyloid-β was pre-treated with 80%formic acid, whereas tau slides needed no pre-treatment. Slides were rinsed in PBS twice for 5 min each before being incubated in primary antibody in a humid chamber at 4°C, overnight.
After primary antibody incubation, slides were rinsed twice in PBS. TH and amyloid-β slides utilized the BioGenex Super Sensitive Kit secondary detection system according to the manufacturer’s protocol: slides were incubated in a SuperEnhancer for 20 min, rinsed in PBS twice, incubated in polymer-HRP for 30 min before being rinsed in PBS and visualized using 3,3’-diaminobenzidine for 9 mins. α-synuclein and tau slides utilized the ImmPRESS kit secondary detection system according to the manufacturers protocol: slides were incubated in the provided secondary antibody for 45 min before being rinsed in PBS and visualized using 3,3’-diaminobenzidine for 5 min. All slides were counterstained using Mayer’s Haematoxylin (VWR) for 1 min, rinsed in running tap H2O, dehydrated in ascending concentrations of ethanol, cleared in xylene and cover-slipped using distrene-plasticiser-xylene.
Microscopic analysis of slides
Image capture of LC sections was carried out using an Olympus Vanox microscope attached to a Micropublisher 3.0 camera at 40× magnification. TH-positive neuronal counts were performed, blinded to the clinical diagnosis, using the manual tag function on Image Pro Plus 7.0. Structures were included in the count if they exhibited a clear neuronal morphology with a defined nucleus and nucleolus. α-synuclein, tau and amyloid pathology were assessed with %-coverage of staining in a 200× objective lens field. This was performed by first flattening the standard color image, then converting it to monochrome. In this mode, pathologic inclusions appeared black and neuromelanin appeared light grey. Thresholds were set based on color intensity to exclude neuromelanin from the capture. %-coverage based on the threshold was then measured (Supplementary Figure 1).
Statistical analysis
Statistical analyses were carried out using GraphPad Prism 7.01. Comparisons of TH, α-synuclein and tau were performed using Mann-Whitney tests for single comparisons, or Kruskal-Wallis tests with Dunn’s post hoc corrections for multiple comparisons. Statistical significance was set at p < 0.05. Intra-class correlations for intra-rater reliability of the %-coverage analyses, and multivariate regression analyses for contributions of demographic factors, were calculated using IBM SPSS 24. Multicollinearity was ruled out with correlation matrices. Though a battery of normality tests were used (Anderson-Darling, D’Agostino-Pearson, Kolgorov-Smirnov, and Shapiro-Wilk tests), we concluded that our large sample size was sufficient to satisfy the central limit theorem, which assumes that the sampling distribution of the mean of any independent random variable will be approximately normal if the sample size is large enough. Coefficients were recorded as coefficient (95%confidence interval) to indicate directionality of the significantly predictive variables in the regression analyses.
RESULTS
Cohort demographics
Demographic analysis of the cohort (Table 1) revealed that the groups were separable by sex, disease duration and Braak α-synuclein stage. The dementia groups had a stark male sex bias compared to the other groups, in line with previous reports [31, 32]. DLB is much more likely to affect males than females with a 2.91 odds ratio found in a previous study [33]. DLB also had a reduced disease duration which was expected because the PDD group had parkinsonism for approximately 10 years on average before the development of a dementia syndrome. DLB also has a reduced age of death due to dementia being a significant cause of mortality in the population. However, DLB cases had a significantly longer duration of dementia than PDD cases. PD without cognitive impairment had a significantly lower mean Braak α-synuclein stage than PDD and DLB, reflecting the greater neocortical pathology found in dementia cases than non-dementia cases in previous reports [17, 18]. Further, Braak tau stage was significantly different between the disease groups with PDD exhibiting the highest mean Braak tau stage. There was comparable tau burden between the other disease groups. Amyloid-β pathology was also highest in PDD, significantly so when compared to PD. In summary, PDD cases showed the highest degree of Alzheimer’s pathology, perhaps highlighting DLB as a dementia syndrome more purely associated with α-synuclein.
Cohort demographics for controls, PD without cognitive impairment (PD), PDD, and DLB
χChi squared analysis performed; KKruskal-Wallis Test performed; MMann-Whitney U Test performed. p < 0.05 indicates statistically significant difference between groups.
LC degeneration in PD and LBD
The quantity of TH + neurons in the LC was significantly reduced in synucleinopathy cases (PD, PDD, and DLB combined) compared to controls (p < 0.0001). When separated into PD, PDD, and DLB, there was a stepwise reduction in LC TH + neuron count with significant differences found between controls and PDD, controls and DLB, and PD and DLB (Fig. 1A, B). The effect of disease phenotype on TH + neuron count prevailed after correction for disease duration, years of dementia, sex, Braak α-synuclein stage, Braak tau stage and in a multivariate regression analysis (coefficient = –11.60 (–17.41 ––5.79) p = 0.0002) which was enhanced by the fact that DLB patients had a shorter disease duration (Supplementary Table 1). In the overall cohort, prolonged disease duration (coefficient = –1.32 (–2.17 ––0.48) p = 0.0024) and increased Braak α-synuclein stage (coefficient = –7.68 (–14.16 ––1.19) p = 0.021) were also significant predictors of LC TH + neuron count, whereas Braak tau stage and Thal amyloid-β phase showed no relationship.
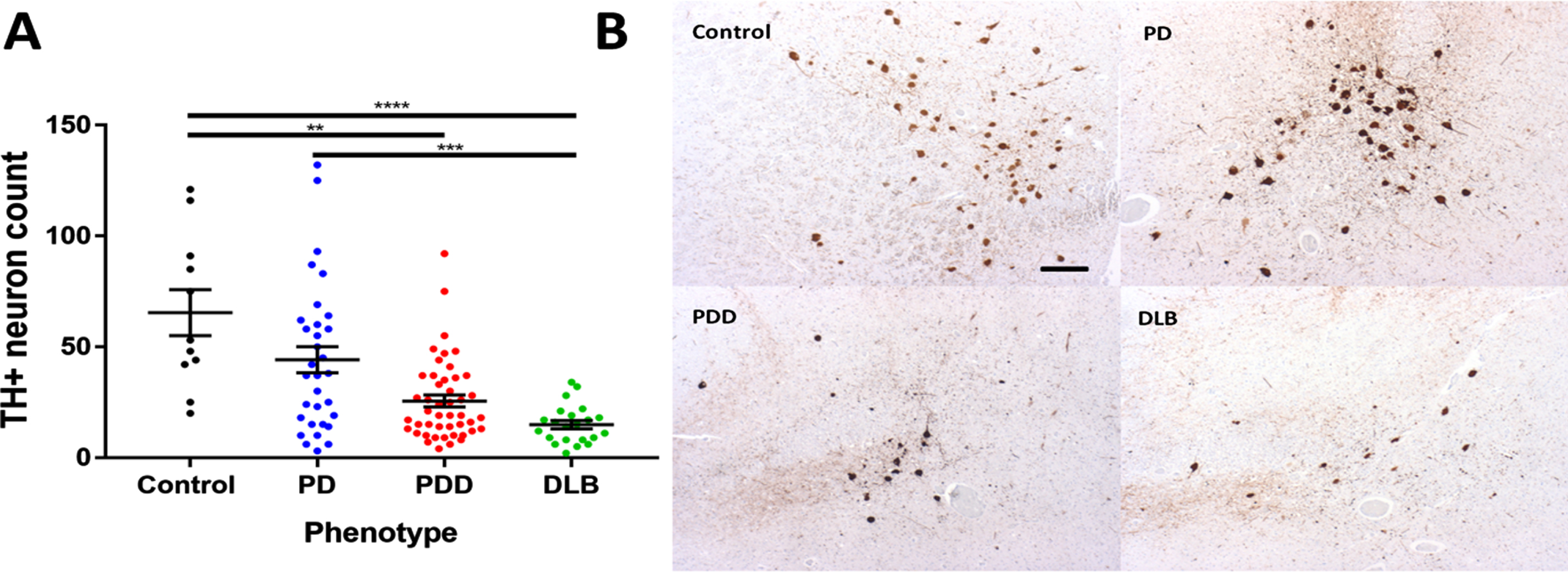
A) TH + neuron counts in the LC in cognitive phenotypes of PD and controls. Statistical analysis was performed by Kruskal-Wallis Test with post-hoc Dunn’s correction for multiple comparisons. **p < 0.01, ***p < 0.001, ****p < 0.0001. Cronbach’s α test = 0.903 indicating high intra-rater reliability. B) Representative photomicrographs of TH-immunostained sections containing the LC in cognitive phenotypes of PD and controls. Images captured under a 4× objective lens. Scale bar in Control panel represents 100μm.
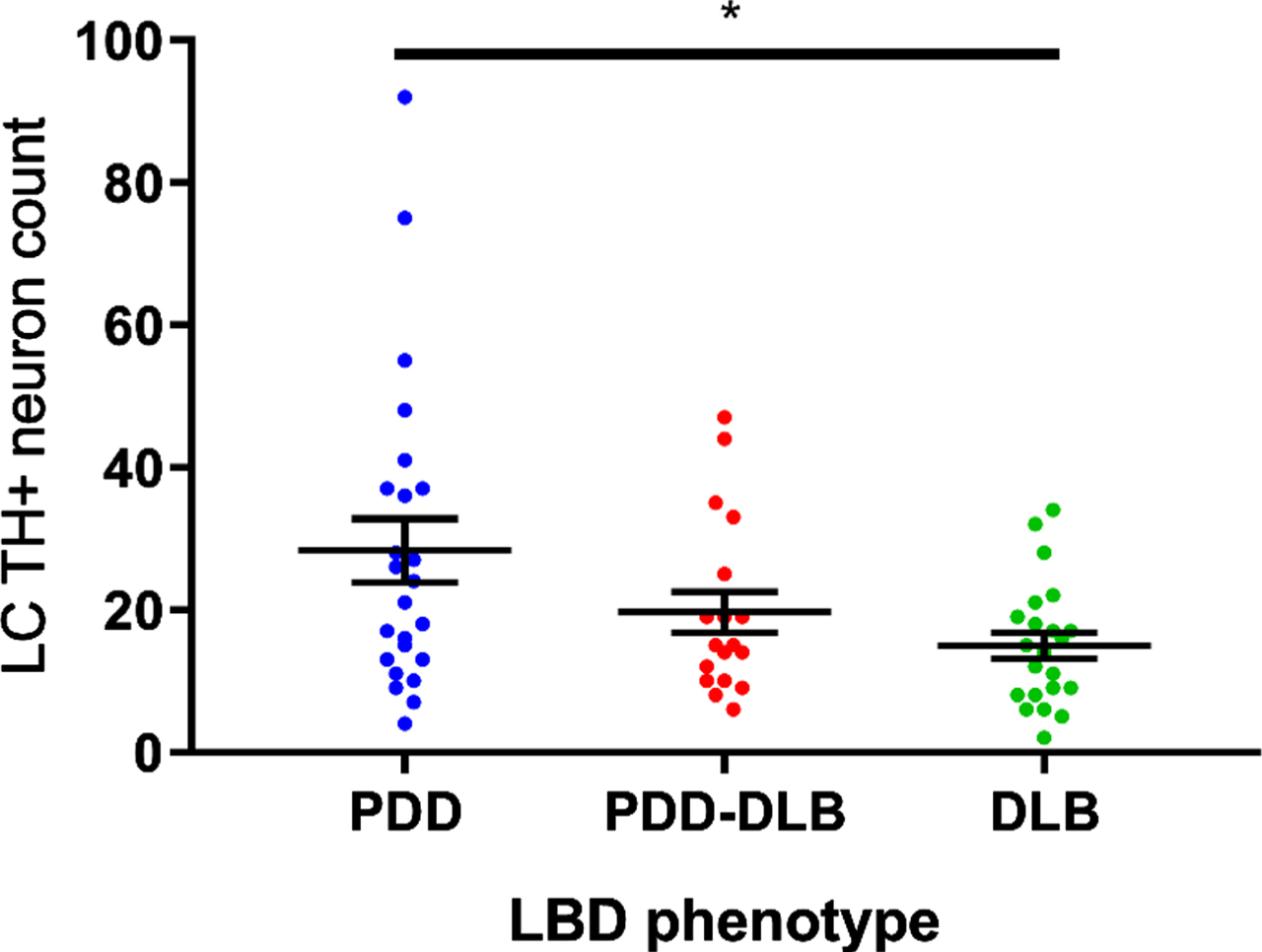
LC integrity in PDD, PDD-DLB and DLB cases. Statistical analysis performed by Kruskal-Wallis Test with post-hoc Dunn’s correction for multiple comparisons: *p < 0.05.
Clinical overlap between PDD and DLB, mirrored by degeneration of the LC
A specific subset of PDD cases show FC and RBD and thus exhibit characteristic features of the DLB-related clinical syndrome (PDD-DLB). Of the 56 PDD cases, 26 showed presence of RBD, VH and FC and were assigned to the PDD-DLB group. TH + neuronal counts in the LC were reanalyzed, accounting for the subdivision of LBD cases into PDD, PDD-DLB and DLB (Fig. 3). This revealed that the PDD-DLB group represented a middle ground between PDD and DLB in terms of LC integrity. These data support the notion in recent literature that LBD represents a continuum with PDD at one end and DLB at the other [34].
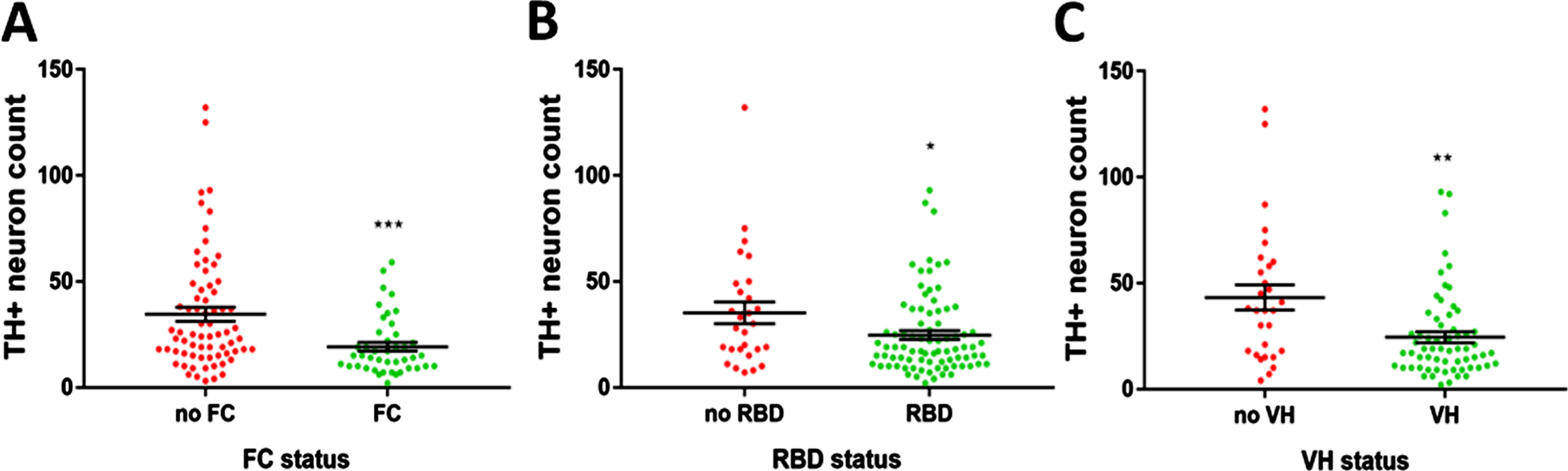
LC integrity relating to the different symptoms of DLB: FC, RBD, and VH. Cases were separated based on the presence or absence of each symptom in the case history in all cases, irrespective of diagnosis. A) The presence of FC. B) The presence of RBD. C) The presence of VH. Statistical analysis performed by Mann-Whitney U test. *p < 0.05, **p < 0.01, ***p < 0.001.
The contribution of LC degeneration to the symptomatology of DLB
Because of the functional role of the LC in controlling the sleep-wake cycle and alert cognitive state, the degeneration of the LC neuron population was assessed for the contribution to each of the three core DLB features—FC, RBD, and VH [5]. VH and RBD were occasionally present in the PD group. The presence of each of these three clinical features, irrespective of overall diagnosis, was associated with significantly reduced LC TH + neuron count (Fig. 3A-C), suggesting that the LC is a significant arbiter of the clinical phenotype. When corrected demographic factors (Supplementary Table 2), only presence of FC (coefficient = –13.41 (–24.07 to –2.761), p = 0.0142) and presence of VH (coefficient = –11.55 (–21.90 to –1.196), p = 0.0292) significantly predicted LC neuron count—the effect of RBD disappeared. There were no significant differences found between depressed and non-depressed groups, and anxious and non-anxious groups.
α-synuclein, tau, and amyloid pathology in the LC in LBD
Using %-coverage methodology, Lewy pathology was found covering the LC field at percentages between 0.3%and 5.3%. Overall, however, there were no discernible differences in the %-coverage of α-synuclein pathology in the LC between, PD, PDD, and DLB, and between LBD subtypes, though DLB showed a significant reduction compared to PDD which disappeared after correction for multiple comparisons (Fig. 4A, B). Lewy pathology was predominantly found intracellularly. Therefore, reduced cell count correlated with reduced overall pathologic %-coverage (r = 0.2634, p = 0.022), By comparison, tau pathology did not correlate with cell count (r = 0.1332, p = 0.248). By multivariate regression analysis, there were no variables (age of death, disease duration, years of dementia, sex, LC TH + neuron count, cognitive group, Braak α-synuclein, %-coverage of tau) that significantly predicted %-coverage of α-synuclein. On the other hand, PDD and DLB were differentiable by the severity of the presence of tau pathology in the forms of neuropil threads and neurofibrillary tangles. PDD cases were significantly more affected by this pathology than DLB cases (Fig. 4C, D). The significant difference in tau pathologic burden was maintained when controlled for disease duration, years of dementia, age of death, sex, %-coverage of α-synuclein, Braak tau stage and TH + cell count (coefficient = –0.2043 (–0.3745 to –0.03410), p = 0.0199; Supplementary Table 3). Amyloid-β granular intracellular deposits were seen in the majority of cases without any discernible variation between groups. In summary, DLB cases appear to display less tau pathology than PDD cases. This indicates that DLB represents a purer synucleinopathy in the LC than PDD.
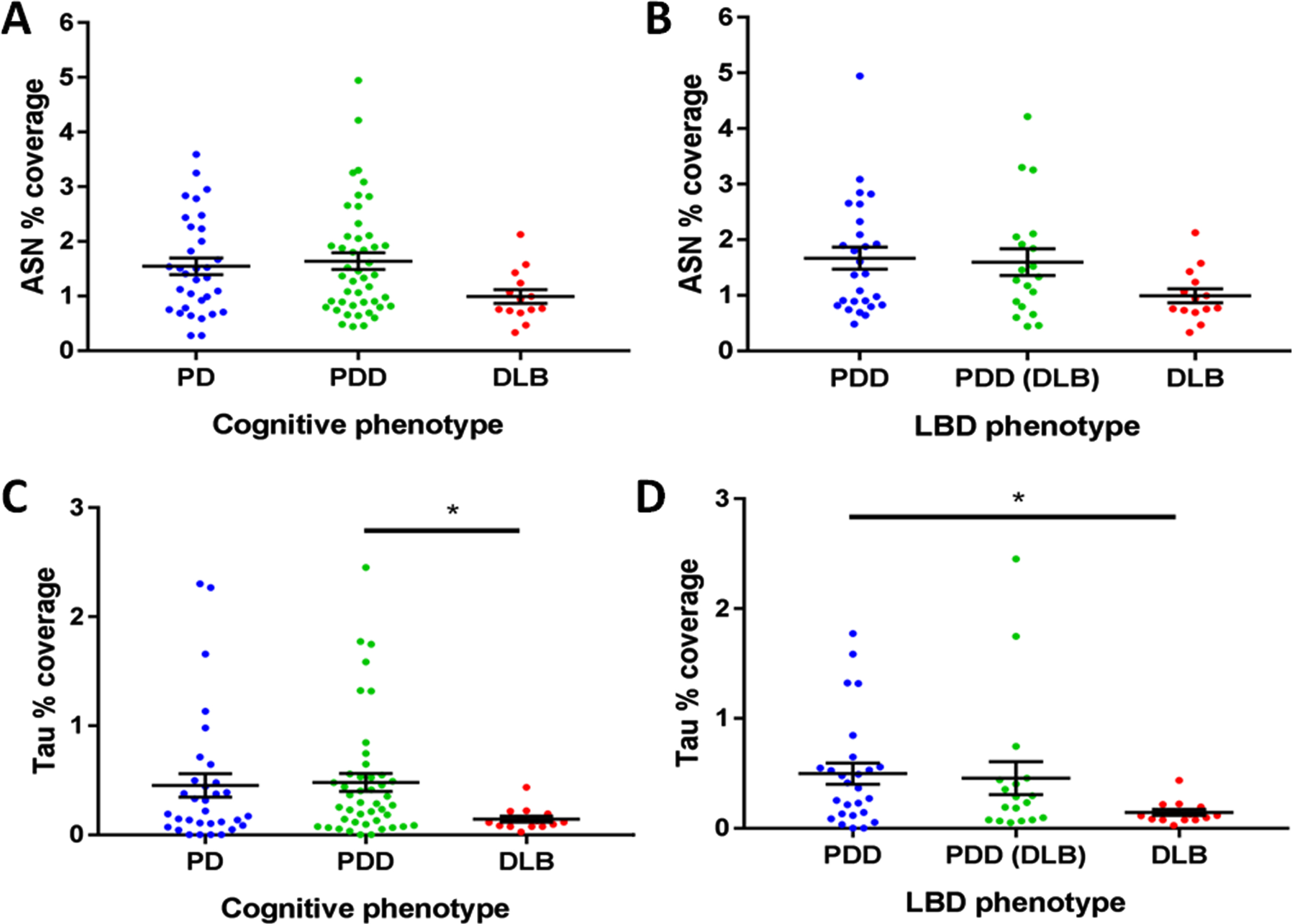
α-synuclein (ASN) and tau pathologic burdens in the LC in cognitive phenotypes of PD and LBD. A) ASN pathologic burden in PD cognitive phenotypes. B) ASN pathologic burden in LBD phenotypes. C) Tau pathologic burden in PD cognitive phenotypes. Statistical analysis was performed with Kruskal-Wallis tests with Dunn’s corrections for multiple comparisons. *p < 0.05. A) PDD vs DLB, p = 0.0219 (before multiple comparisons). B) PDD vs DLB, p = 0.0237 (before multiple comparisons).
DISCUSSION
The most important finding of the present study is the exhibition of pathological changes in the LC that differentiate between subtypes of LBD. The differential burden of pathology across LBD cases illustrates a continuum whereby DLB is more severely affected than PDD. The difference between LC counts in these groups are made even more stark by the fact that PDD cases have a longer disease duration. This suggests that the LC undergoes significantly more degeneration in DLB, despite having lower disease duration. PDD cases that exhibited DLB-related symptoms showed a level of pathology intermediate to PDD and DLB. The contribution of the LC to the dementia syndrome in these cases may relate to DLB-related symptoms such as FC, RBD, and VH. However, the significance of RBD status disappeared when corrected for demographic variables. We have shown that the presence of these symptoms individually is associated with reduced LC neuron count, though further work will need to be performed to elucidate the role of the LC in producing these symptoms. From this study, it can be hypothesized that the LC contributes more to the dementia syndrome in LBD than dementias of differing etiologies. This was highlighted in previous studies where it was found that LBD had significantly more degeneration of the LC than in cases of vascular or frontotemporal lobar dementia [35, 36]. Interestingly, these studies did not investigate the deposition of pathologic proteins in their cohorts, nor did they separate LBD into DLB and PDD as we have in the present study. The relationship between LC neuron count and pathologic burden was the inverse of what might have been expected, with more pathology being present in more intact neuronal populations. Deposition of protein aggregates occurring intracellularly resulted in higher %-coverage of pathology in more intact neuronal populations. Interestingly, there was no predictive relationship between the burden of α-synuclein and tau pathology in the LC, indicating that co-pathology was unlikely to have driven the differences between disease groups. Of further intrigue are the apparent discrepancies between subcortical and cortical pathology in differentiating PDD and DLB. Previous studies have shown that the majority DLB exhibits a higher burden of AD co-pathology in the cortex, basal ganglia and parahippocampal regions than PDD [37–39]. In our study, we have demonstrated that there is minimal brainstem AD pathology in DLB, with a comparatively higher amount in PDD cases. Our PDD and DLB cases have comparable Thal amyloid-β phases.
Our results indicate a noradrenergic deficit in PD that particularly affects LBD cases. The role of noradrenaline in the pathophysiology of PD has been the subject of increased focus in recent literature [40–42]. We have been able to show that the noradrenergic deficit in LBD might be a distinguishing feature compared to PD cases without cognitive impairment. Moreover, we have shown that noradrenaline has the potential to control the nature of the dementia phenotype present in LBD cases whereby the presence of FC, RBD, and VH are associated with evidence of a stronger noradrenergic deficit than those where these features are not present. However, when corrected for demographic and disease-related factors, the effect of RBD presence on LC TH + neuron count disappeared, bringing our study in line with previous studies on the integrity of the post-mortem LC in LBD with or without RBD [43]. However, in vivo evidence for the role of the LC in these symptoms has also been found using T1-weighted neuromelanin-sensitive MR imaging. A study by Garcia-Lorenzo and colleagues found that PD patients with RBD had reduced signal in the LC compared to those without RBD and controls [44]. This lends support to the notion that LBD exists on a clinical and pathological continuum [32, 34], and that LBD pathology should be thought about in terms of the clinical symptoms present rather than the timing of dementia relative to the onset of motor symptoms.
The mesopontine reticular formation and coeruleus/subcoeruleus regions are critical for the regulation of the sleep-wake-cycle. The harbinger of REM-sleep is the onset of specific forms of PGO waves, which typically occurs 30–90 s before REM-sleep [24]. PGO waves are initiated and regulated by the coeruleus/subcoeruleus region and mesopontine reticular formation [45]. These regions have crucial roles in sleep-wake-cycle regulation by performing the switching of thalamic relay nuclei from the waking relay mode, where they transmit sensory signals to the cortex, to the sleeping burst mode where they do not transmit these signals [25]. Alterations of PGO signals may cause “sleep disturbance and abnormal cortical processing of sensory afferents.” [25] Therefore, it is possible to think of hallucinations as abnormal encroachment of sleep characteristics into the waking state, i.e., dreams. The pattern of symptoms present in DLB—VH, RBD, and FC—paint a picture of sleep-wake-cycle dysregulation whereby VH represents dreams encroaching on the waking state, RBD represents inappropriate motor activity during ‘deep sleep’ and FC represents variations in arousal. Unresponsive periods, and hypersomnolent episodes are also frequent in DLB and form some of the supportive features of a DLB diagnosis [5]. Severe pathology of the coeruleus/subcoeruleus region and mesopontine reticular formation, resulting in dysregulation of the sleep-wake-cycle could plausibly underlie all of these clinical DLB-related phenomena. Given that our study found involvement of the LC in the incidence of VH and FC, it would be interesting for further studies to assess the subcoeruleus and reticular formation for their contributions to DLB symptomatology.
Identification of the pathologic substrates of specific dementia symptoms would allow for more tailored treatments of an individual’s unique dementia syndrome. Currently, the only licensed drug class LBD are the cholinesterase inhibitors, thought to act by addressing the cortical cholinergic deficit caused by loss of neurons in the cholinergic basal forebrain [46]. Noradrenergic fibers originating from the LC have been shown to innervate the nucleus basalis of Meynert [47] which is one of the four main basal forebrain cholinergic nuclei and has been shown to exhibit cell loss in LBD [48]. A loss of noradrenergic innervation in this area early in the disease course may underlie nucleus basalis degeneration and the resultant cortical cholinergic deficit. Noradrenergic medications have also been shown to improve certain aspects of cognition in PD as well as improving other neuropsychiatric symptoms such as impulsivity, anxiety and depression [42]. Noradrenaline has been shown to induce long-term potentiation of hippocampal synapses by acting at β-adrenergic receptors [49]. Receptor binding studies in this area have shown downregulation of key adrenergic receptors in DLB cases [50].
Our study is not without limitations, including the retrospective nature of our case history analysis and sampling the LC at only one rostro-caudal level. Another potential limitation is the use of representative sections of the LC instead of unbiased stereological methods to perform neuron counts. However, previous studies using single representative section counts and stereological estimates have found a high degree of comparability [51]. Multivariate regression analysis was used to assess the contribution of other variables to the LC TH + neuron counts, despite the fact this variable did not pass normality testing. Though the central limit theorem states that the larger the sample size the higher the chance that a normal distribution can accurately be assumed, the findings should be assessed in the context of not having passed normality tests. Nonetheless, our findings have highlighted an important aspect of the pathophysiology of LBD which may have implications for the administration of noradrenergic medications in clinical practice. Future work will involve studies of the cerebral targets of LC-noradrenergic system, including the amygdala, hippocampus and cerebral cortex. In vivo neuroimaging studies should focus on the LC-noradrenergic system to validate our neuropathologic data during life. Clinical trials of the effects of noradrenergic drugs on specific dementia symptoms in LBD are warranted.
In conclusion, the LC-noradrenergic system is significantly affected in PD and LBD specifically. Deficits in this system, resulting from neuronal loss and dysfunction of surviving neurons, may underlie the onset of DLB-related symptoms such as RBD and FC. Based on a symptom-by-symptom approach, we have provided further evidence that LBD is more likely to reflect a continuum caused by different severity of pathology in several key brain regions including the LC. Further work will be needed to validate these data in vivo, and to assess the clinical efficacy of noradrenergic drugs as an adjunct or alternative to cholinesterase inhibitors in the treatment of LBD.
Footnotes
ACKNOWLEDGMENTS
We would like to give special thanks to the donors and their families who donated their brains to the Parkinson’s UK Tissue Bank. This work was funded by the Jean Shanks Foundation as part of Bension Tilley’s PhD studies and the National Institute of Health National Institute on Aging, Arkansas, USA: grant no. NIA AG12411.
CONFLICT OF INTEREST
The authors have no conflicts of interest to report.