
Abstract
The question whether Parkinson’s disease dementia (PDD) and dementia with Lewy bodies (DLB) are expressions of the same underlying disease has been vigorously debated for decades. The recently proposed biological definitions of Lewy body disease, which do not assign any particular importance to the dopamine system over other degenerating neurotransmitter systems, has once more brought the discussion about different types of Lewy body disease to the forefront. Here, we briefly compare PDD and DLB in terms of their symptoms, imaging findings, and neuropathology, ultimately finding them to be indistinguishable. We then present a conceptual framework to demonstrate how one can view different clinical syndromes as manifestations of a shared underlying Lewy body disease. Early Parkinson’s disease, isolated RBD, pure autonomic failure and other autonomic symptoms, and perhaps even psychiatric symptoms, represent diverse manifestations of the initial clinical stages of Lewy body disease. They are characterized by heterogeneous and comparatively limited neuronal dysfunction and damage. In contrast, Lewy body dementia, an encompassing term for both PDD and DLB, represents a more uniform and advanced stage of the disease. Patients in this category display extensive and severe Lewy pathology, frequently accompanied by co-existing pathologies, as well as multi-system neuronal dysfunction and degeneration. Thus, we propose that Lewy body disease should be viewed as a single encompassing disease entity. Phenotypic variance is caused by the presence of individual risk factors, disease mechanisms, and co-pathologies. Distinct subtypes of Lewy body disease can therefore be defined by subtype-specific disease mechanisms or biomarkers.
INTRODUCTION
The question whether Parkinson’s disease (PD) and dementia with Lewy bodies (DLB) are expressions of the same underlying disease has been vigorously debated ever since consensus diagnostic criteria for DLB were first proposed in 1996 [1–7]. However, the need to resolve this discussion has not diminished in importance, given that the international research community is actively working towards a biological redefinition of various Lewy body diseases [8, 9].
Here, we review the literature on differences and similarities between DLB and PD dementia (PDD), including clinical symptoms, imaging findings, neuropathology, and the importance of co-pathologies. We find that PDD and DLB are, for all practical purposes, indistinguishable disease entities, reflective of an advanced stage of Lewy body disease often accompanied by Alzheimer disease (AD) co-pathology and widespread neurodegeneration.
To illustrate the problem of clinically-based PD and DLB diagnoses, Table 1 summarizes key information from three typical cases. Two of these patients, who are assigned the different diagnostic labels of DLB and PD, are virtually identical in every single aspect except for a slight difference in the motor-dementia interval. In contrast, both of these patients are strikingly different from the third patient, who nevertheless is also diagnosed with PD.
Three typical patients with Lewy body disease
Characteristics of three patients with Lewy body disease. Patient 1 (DLB) and patient 2 (PD) show virtually identical findings but both are very different from patient 3 (PD).
Rather than upholding several distinct diagnoses based on separate clinical diagnostic criteria, we argue for the validity of a biological definition of Lewy body disease and its possible subtypes supported by biomarkers. The well-known clinical syndromes can thereby all be encompassed under an umbrella term of Lewy body disease. Some of these clinical syndromes, including early PD, isolated REM sleep behavior disorder (iRBD), pure autonomic failure (PAF), and idiopathic hyposmia, will reflect relatively restricted α-synuclein associated neurodegeneration. In contrast, clinical PDD and DLB will typically be evidence of more extensive and severe αSyn-associated neurodegeneration affecting multiple neurotransmitter systems. In the recently proposed staging systems, a patient will be diagnosed based on the presence of pathogenic, aggregated αSyn and neurodegeneration known to be specifically associated with this pathology [8, 9]. Other biomarkers may serve to define biological subtypes.
In the following, we shall briefly compare PDD and DLB with respect to symptoms, imaging findings, and neuropathology. We then propose a framework to illustrate how it is possible to think about different clinical syndromes as manifestations of the same underlying Lewy body disease. We also emphasize the importance of individual risk factors and co-pathologies, especially AD co-pathology, which seems to work synergistically with or additively to Lewy pathology to shape symptomatology in individual patients.
COGNITIVE AND PSYCHIATRIC SYMPTOMS
The cognitive and neuropsychiatric symptoms seen in DLB and PDD seem to be indistinguishable [10, 11]. Cognitive deficits in both disorders occur across all cognitive domains, including impairments in executive abilities, working memory, attention, visuospatial skills, processing speed and language [12–23]. It is controversial if memory is less affected in the early disease [24–26].
Neuropsychiatric symptoms are also very common in both PDD and DLB [27]. A systematic review [10] summarized the frequencies of neuropsychiatric symptoms in PDD and DLB as follows: depression (PDD 58%, DLB 49%), apathy (PDD 54%, DLB 57%), anxiety (PDD 49%, DLB 65%), hallucinations (PDD 54%, DLB 76%) and delusions (PDD 29%, DLB 57%) [27–29].
In general, these studies find that the frequency of cognitive and neuropsychiatric symptoms is very similar in PDD and DLB, or sometimes slightly higher in DLB. Thus, many individual PDD patients are indistinguishable from DLB patients. A recent large longitudinal study also demonstrated that diagnosed PDD and DLB patients show virtually identical rates of cognitive and motor decline when followed for up to five years [30]. Thus, based on the cognitive and neuropsychiatric profiles, there appears to be no justification to assume that PDD and DLB are different diseases.
IMAGING BIOMARKERS
Radiotracer and MRI studies generally report nearly identical findings in PDD and DLB. Very similar patterns of cerebral hypoperfusion and hypometabolism have been reported, with the most pronounced deficits seen in posterior parieto-occipital cortices [31–33].
Structural MRI studies of PDD and DLB have revealed comparable patterns of mostly posterior atrophy, occasionally slightly more pronounced in DLB [34, 35]. However, any statistical differences between PDD and DLB is only apparent when comparing large groups of patients, and many individual PDD and DLB patients are indistinguishable. Of note, atrophy will often be aggravated by the presence of co-morbid AD pathology, which is more common in DLB [36–38].
PET studies have shown significant depletion of cholinergic projections from the basal forebrain to the cortex in both PDD and DLB, with the most notable effects observed in the posterior parieto-occipital regions. Again, there is no obvious differences in the regional pattern or magnitude of loss in DLB compared with PDD [32, 39–43].
Patients with PD and DLB show similar loss of nigrostriatal dopaminergic innervation on dopamine transporter (DAT) striatal imaging [32, 45]. When compared to non-demented PD patients, DLB shows considerably more symmetric loss and also more uniform loss in both the putamen and caudate nucleus [44, 45]. However, it is known that the dopaminergic deficit in PD becomes more symmetric at follow-up and also with progressive involvement of the caudate [46]. Thus, as non-demented PD patients progress, their dopamine scans look increasingly like DLB patients.
A small percentage (10–15%) of clinically-diagnosed DLB patients exhibit relatively normal DAT scans at diagnosis [47]. However, one study showed that when DLB patients with normal baseline DAT imaging were reexamined 3 years later, they had all progressed and showed pathologically low dopamine innervation [48]. A recent longitudinal study also demonstrated that patients with a baseline DLB diagnosis showed faster motor progression over an 8-year period compared to patients with a baseline PDD diagnosis [49].
Lewy body disease is characterized by profound loss of cardiac sympathetic innervation, which can be visualized with [123I]MIBG or [18F]fluorodopamine scans [50, 51]. At diagnosis, many RBD-negative PD patients display normal or near-normal MIBG parameters, whereas virtually all de novo PD patients with RBD show markedly pathological MIBG scans [52]. However, once PD patients have progressed to Hoehn & Yahr stage III, they almost always exhibit severe sympathetic denervation on MIBG [53, 54]. Likewise, nearly all DLB patients show pathological MIBG scans already at diagnosis [55–57]. Put together, virtually all patients with Lewy body disease will eventually develop severely pathological MIBG scans as the disease progresses, with no apparent differences between PDD and DLB.
In summary, many individual PDD and DLB patients have very similar, or identical, findings on all the aforementioned imaging modalities. At the group level, DLB patients sometimes show more severe pathology on imaging, but a large percentage of individual PDD and DLB patients are identical.
One notable exception relates to the frequency of AD co-pathology in PDD vs. DLB. A meta-analysis of amyloid PET scans concluded that 68% of DLB, 34% of PDD, and 5% of non-demented PD patients had pathological findings on amyloid PET [58, 59]. The implications of this important observation are discussed in more detail below.
NEUROPATHOLOGY
When comparing PDD and DLB, neuropathologists generally conclude that no pathologic substrate found at postmortem can reliably differentiate these clinically-defined disorders [6, 60–62]. Commonly, PDD patients show higher levels of neocortical Lewy pathology compared to non-demented PD patients, signifying that PDD represents a later, more advanced disease stage than non-demented PD [63, 64]. On the other hand, PDD and DLB patients show very similar global distribution and severity of Lewy pathology, and at autopsy the vast majority of cases are Braak Lewy body stage 5–6 [11, 66]. Thus, with respect to Lewy body pathology there are no obvious differences between PDD and DLB.
Importantly, most studies report that intermediate-to-high levels of AD co-pathology (i.e., neurofibrillary tangles and beta-amyloid plaques), are more common in DLB than in PDD [60, 68], although some have reported equal levels [66]. Many studies have reported a strong association between AD co-pathology and reduced cognitive status in autopsied PD cases [63, 69–73]. Generally, a combination of Lewy and AD pathologies shows a stronger correlation with dementia compared with patients with pure Lewy body disease [60, 74]. As mentioned above, a similar relationship is seen with amyloid PET scans, which are positive in around 2/3 of DLB patients, 1/3 of PDD, but in only 5% of non-demented PD [58, 59].
Of note, at least two studies have clearly shown that the motor-dementia interval is strongly influenced by the amount of AD co-pathology (Fig. 1) [60, 67]. Demented patients with no or low-level AD co-pathology at postmortem usually develop parkinsonism many years before dementia. In contrast, patients who develop dementia within a few years of parkinsonism (irrespective of whether they had a PDD or DLB diagnosis clinically) more often have intermediate- or high-levels of AD pathology.
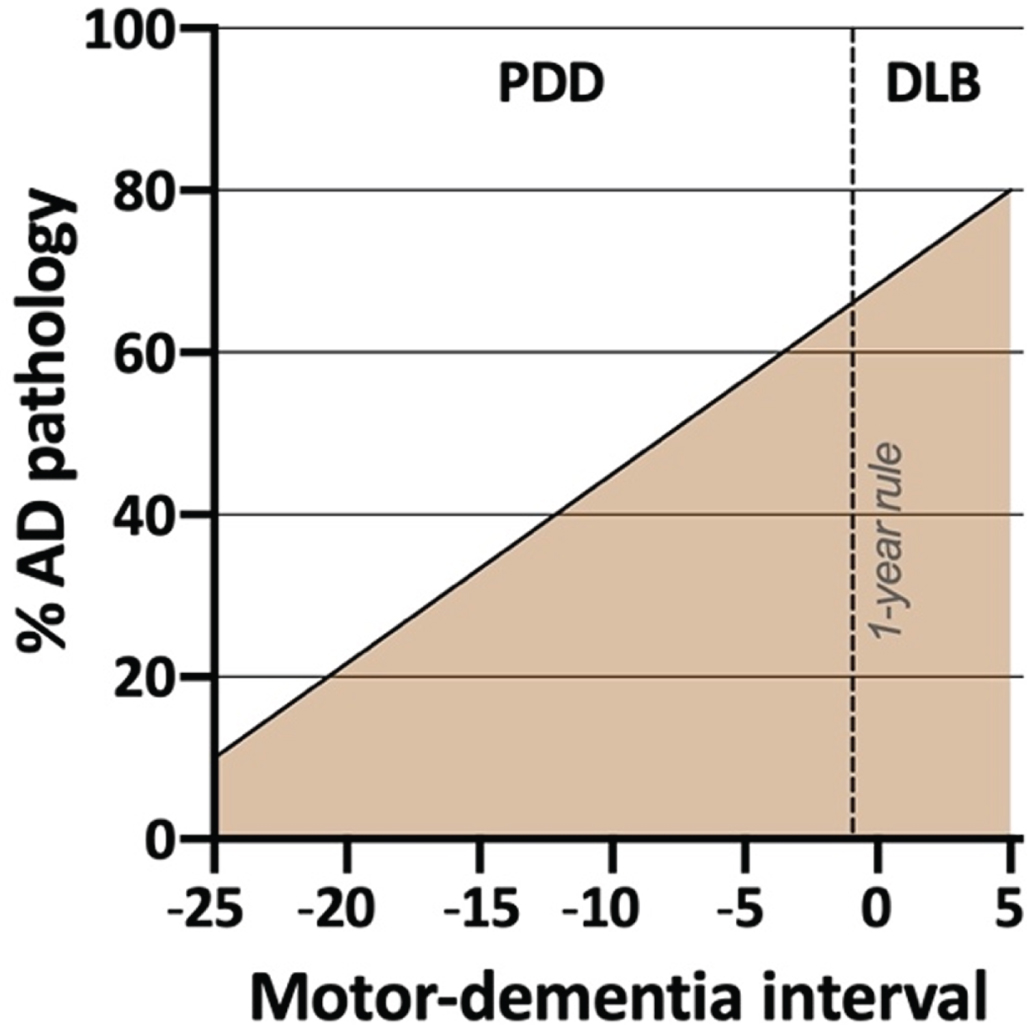
Schematic illustration of the prevalence of significant AD co-pathology at postmortem in individuals with Lewy body dementia. Higher rates are observed in patients with short motor-dementia intervals. For example, a PDD patient with a 5-years motor-dementia may have a 60% probability of AD co-pathology at death, whereas another PDD patient with a 20-years motor-dementia may have only a 20% probability of AD pathology at death. No obvious distinction is evident between DLB and the PDD patients with the shortest motor-dementia interval, underscoring the arbitrary nature of “the 1-year rule” (based on data from [58, 67]).
These postmortem and PET imaging data strongly suggest that the presence of AD co-pathology accelerates cognitive decline. In short, relatively pure Lewy pathology most commonly results in a PD diagnosis, and the development of the advanced PDD stage typically takes many years. The few DLB cases without AD-pathology at postmortem frequently had GBA mutations or other co-pathologies such as cerebrovascular disease [60]. In contrast, significant amounts of AD co-pathology regularly lead to more rapid dementia, but again, there is no clear difference between PDD patients with a short motor-dementia interval and DLB patients with an even shorter motor-dementia interval (Fig. 1). Of note, PDD patients with considerable amounts of AD co-pathology seems to have clinical phenotype that in many respects is more similar to DLB [60, 75–77], and one study showed that the arbitrary “1-year rule” had very poor diagnostic accuracy with respect to predicting the amount of neocortical Lewy bodies or the amount of AD co-pathology in PDD vs. DLB [60].
In summary, postmortem studies show that PDD and DLB have highly similar burdens and distribution of Lewy pathology. Significant AD co-pathology is more commonly seen in DLB and in PDD patients with a short motor-dementia interval, but with no clear distinction between the two.
RELATIONSHIP BETWEEN LEWY BODY DISEASE AND AD CO-PATHOLOGY
Figure 2 provides a visual summary to illustrate how AD and Lewy body pathologies might theoretically interact to produce the different clinical syndromes of DLB, PD and PDD.
The rows in the figure depict Lewy body disease divided clinically into brain-first and body-first disease [52, 79]. The latter is characterized by development of iRBD, autonomic symptoms, and pathological MIBG scintigraphy before diagnosis [52], and more often shows relatively symmetric dopamine degeneration [45, 80]. These symptoms and signs are all risk factors for rapid dementia (summarized in [78]). Thus, body-first Lewy body disease in itself leads to faster cognitive decline compared to RBD-negative brain-first disease.
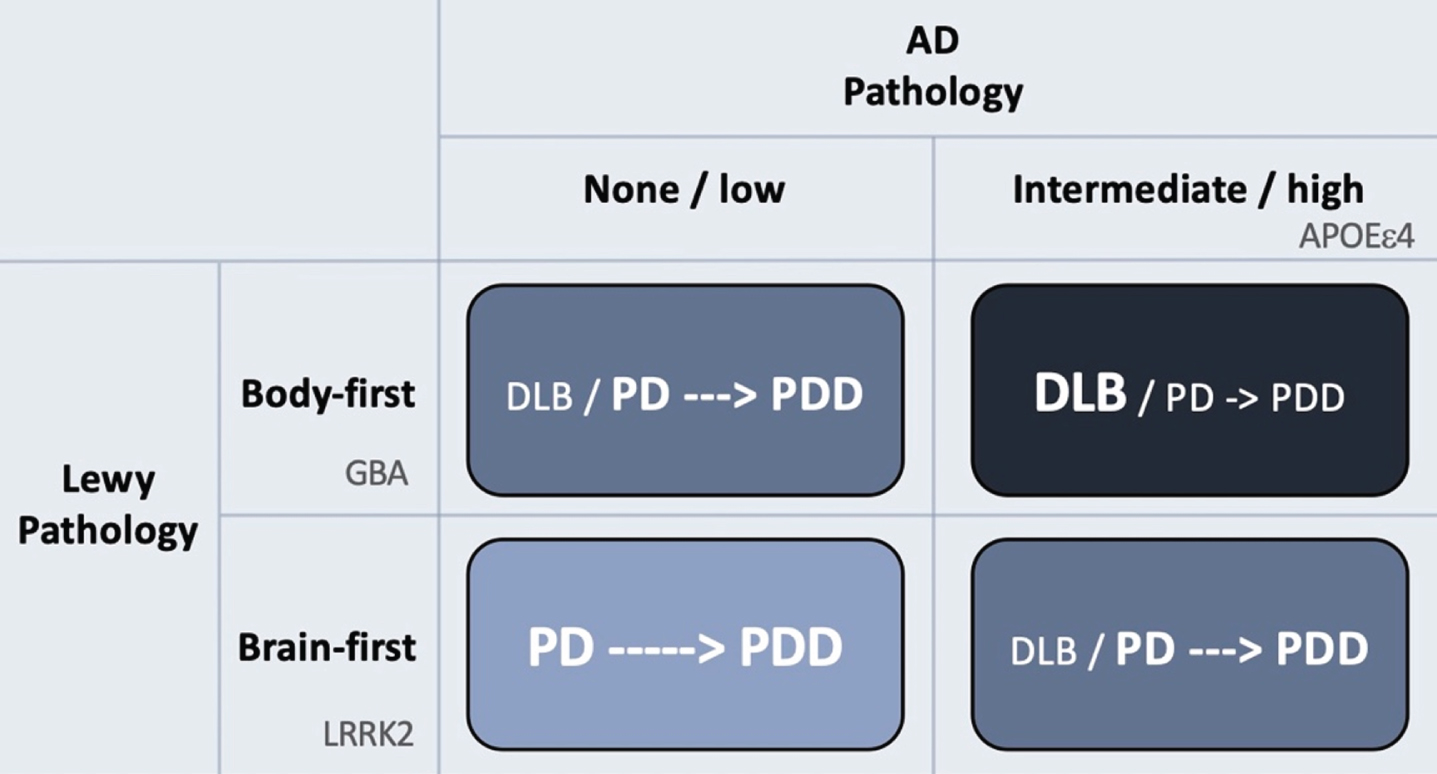
Hypothetical interaction between Lewy body disease and AD-type co-pathology. Patients with clinical body-first Lewy body disease show faster progression towards dementia compared to brain-first disease. Prodromal iRBD, autonomic symptoms, pathological MIBG scintigraphy, and symmetric dopamine degeneration are all risk factors of accelerated cognitive decline and signify a body-first etiology. AD-type co-pathology is also strongly associated with fast dementia in Lewy body disease. Thus, body-first Lewy body disease with co-morbid AD-pathology will often result in a DLB diagnosis or sometimes a PD diagnosis with rapid dementia (dark blue quadrant; short arrow signifies fast progression to dementia). Brain-first Lewy body disease without concomitant AD-pathology is almost always diagnosed as PD and the evolution towards dementia is often much slower (light blue quadrant; long arrow signifies slow progression to dementia). The other remaining quadrants result in clinical syndromes with in-between rates of progression towards cognitive decline. Finally, the figure shows how different genetic risk factors could theoretically be distinctly associated with the different axes of Lewy body vs. AD pathology. LRRK2 mutations are associated with a benign, RBD-negative Lewy body disease, and thus not with dementia or DLB. GBA mutations associate with an RBD-positive, fast progressive Lewy body disease but not with AD. The association between GBA and dementia is therefore related mainly to the axis of Lewy body disease. In contrast, APOEɛ4 promotes the formation of AD co-pathology and is therefore also associated with dementia in Lewy body disease, due to the contribution from AD pathology.
The columns of Fig. 2 depict the burden of AD co-pathology. As reviewed above, AD co-pathology is also strongly associated with faster dementia in Lewy body disease. Hence, it appears that patients diagnosed with DLB mainly consists of individuals, who have a combination of body-first Lewy body disease and co-morbid AD pathology. If such cases receive a PD diagnosis first, they will most likely progress rapidly to PDD.
In contrast, patients with brain-first Lewy body disease in the absence of AD co-pathology nearly always receive a PD diagnosis, and the evolution towards PDD is much slower. Such patients are generally younger, display initially more asymmetric dopamine loss, fewer non-motor symptoms, and are RBD-negative at diagnosis [81–84].
Patients in the remaining two quadrants (Fig. 2) will probably show in-between rates of progression towards cognitive decline.
Finally, it should be remembered that the relationship between AD and Lewy pathology is probably not simply additive. There is evidence to suggest that these pathologies can cross-seed each other [85–87]. Of note, patients with early brainstem-predominant Lewy body disease comprise almost half of all cases in postmortem series, and they do not exhibit more AD co-pathology than the background population. In contrast, patients with advanced Lewy body disease on average display much higher frequencies of AD pathology than expected [58, 88–90]. This observation implies that the gradual accumulation of Lewy pathology can trigger the formation of AD co-pathology.
Given the strong association between AD co-pathologies and cognitive decline in Lewy body disease, it is an exciting prospect that anti-amyloid treatments could potentially slow down cognitive decline in Lewy body disease.
GENETICS
A review of genetic risk factors in Lewy body disease is beyond the scope of this paper, so only a few interesting associations will be highlighted here. Several studies have demonstrated that mutations in the glucocerebrosidase (GBA) gene in PD are associated with a more severe phenotype of Lewy body disease, including faster progression, increased prevalence of RBD and other non-motor symptoms, accelerated cognitive decline, and reduced survival. Not surprisingly, GBA mutations are also associated with the clinical DLB diagnosis [91–96].
In contrast, mutations in the leucine-rich repeat kinase 2 (LRRK2) gene are generally associated with more benign type of Lewy body disease with slower progression, lower risk of RBD and other non-motor manifestations, and late dementia in PD, and are not associated with DLB [97–100].
The apolipoprotein E4 (APOE ɛ4) genotype strongly elevates the risk of AD [101, 102]. Some studies did not detect associations between APOE ɛ4 and elevated risk of either PD or iRBD [103, 104], but do show that this genotype predicts dementia in PD [61, 105–107]. In any case, the APOE ɛ4 genotype seems to be much more strongly associated with AD than with Lewy pathology.
Thus, it can be speculated that different genetic risk factors are mainly associated with independent components of protein pathologies (Fig. 2). LRRK2 mutations might be associated with a relatively benign olfactory-first or brain-first etiology of Lewy body disease and is therefore not associated with dementia or DLB. In contrast, GBA mutations, given their more rapid disease progression and high non-motor symptom comorbidity, may predispose to a body-first etiology of Lewy body disease leading to faster dementia and is therefore associated with DLB. Importantly, GBA mutations are not associated with AD per se, so its association with DLB seems to relate purely to the Lewy body component of DLB. Finally, the APOE ɛ4 genotype is also associated with DLB, not because of any particularly strong association with Lewy body disease, but rather because it promotes concomitant AD pathology, which in itself accelerates cognitive decline.
EVOLUTION OF LEWY BODY DISEASE
Figure 3 depicts a hypothetical framework to illustrate how Lewy body disease may evolve from the preclinical stage, over several heterogenous early- and moderate-stage subtypes, to finally converge on a more homogenous, advanced stage, during which the patient typically becomes demented. The advanced stage can be termed Lewy body dementia and includes both PDD and DLB. Such patients are characterized by widespread deposition of Lewy body pathology in both subcortical and most commonly (but not always) cortical regions. Moreover, multiple neurotransmitter systems, including autonomic, noradrenergic, dopaminergic, serotonergic, and cholinergic, are typically significantly damaged at this clinical stage, and many patients display considerable amounts of comorbid AD pathology. Patients with DLB and PDD have similarly poor prognosis with a median survival from onset of dementia of 4–6 years [108, 109].
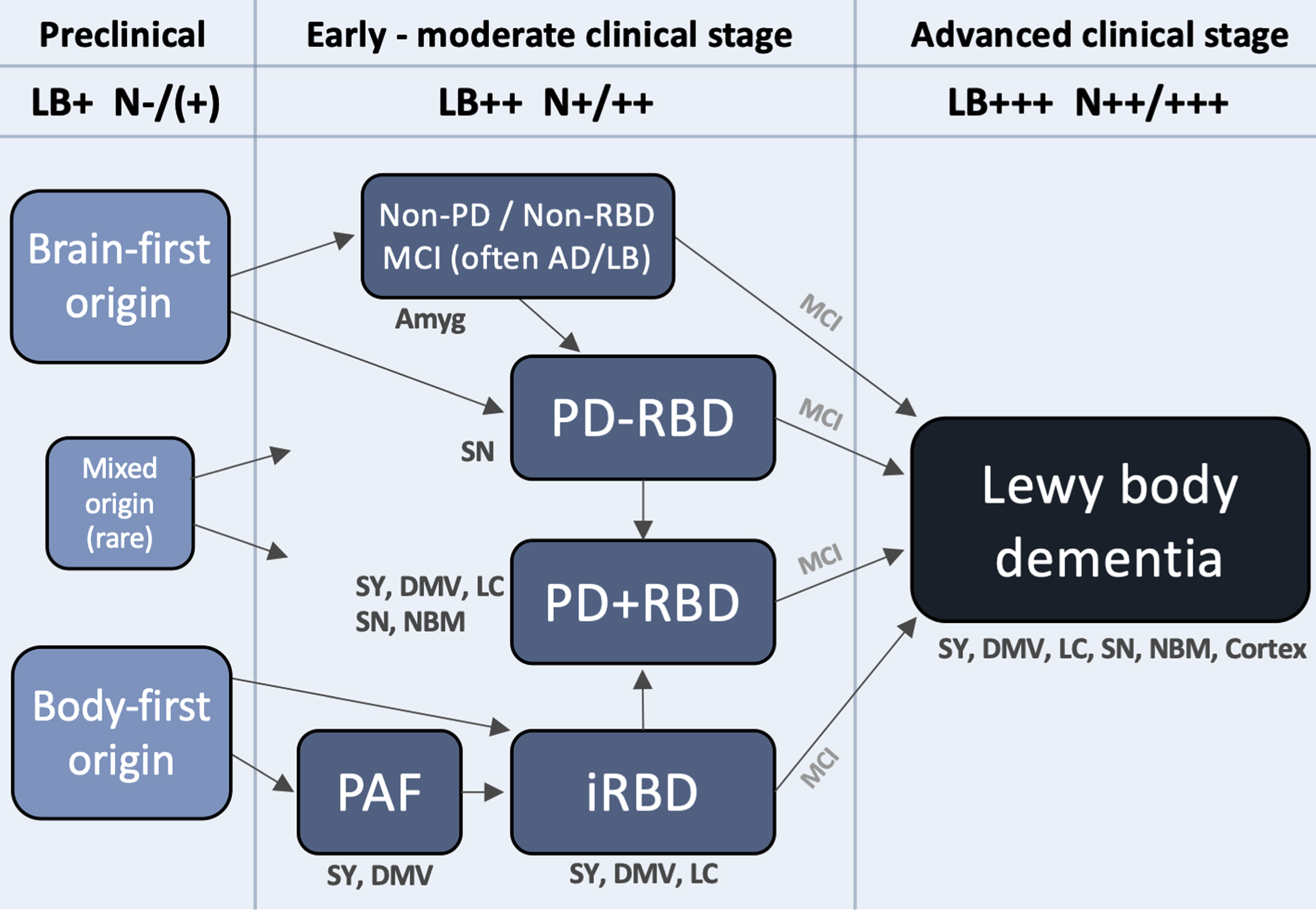
A hypothetical framework to illustrate how Lewy body disease develops from the pre-clinical stage over several possible well-defined early- and moderate clinical disease stages to converge on a more homogenous advanced disease stage, in which patients are typically demented. Rare exceptions to the shown trajectories may exist. The figure lists some neuroanatomical structures, which typically show damage during that particular clinical stage. Based on postmortem data, imaging, and clinical phenotypes, most patients can be categorized into two overall types: brain-first (olfactory/limbic-first) or a body-first (gut/autonomic-first). A small group with mixed (multi-focal) origin of αSyn-pathology may lead to equally mixed clinical symptom subtypes. Irrespective of the origin of αSyn-pathology, and of the heterogeneity seen in symptoms, neurodegeneration, and αSyn deposition during earlier clinical subtypes, patients will tend to converge on a homogenous advanced stage characterized by widespread αSyn pathology and neurodegeneration, parkinsonism, RBD, non-motor symptoms, and cognitive decline. The figure also depicts approximate amounts of Lewy pathology (LB) deposition, and neurodegeneration (N). Pre-clinically, patients are by definition LB-positive but the level of neurodegeneration N, if any, is insufficient to cause symptoms. Conversely, in the advanced stage, widespread LB pathology in peripheral and CNS structures are commonly seen accompanied by marked multi-system neurodegeneration. Amyg, amygdala; DMV, dorsal motor nucleus of vagus; LC, locus coeruleus; MCI, mild cognitive impairment; NBM, nucleus basalis of Meynert; SY, sympathetic ganglia and fibers.
Initially there is a preclinical disease stage in which pathological αSyn may be detectable in CSF, blood, or other tissues, but before significant neurodegeneration has occurred. This is followed by several well-known clinical syndromes, characterized by limited symptoms, and relatively restricted neuronal damage. These syndromes include parkinsonism (including early PD), iRBD, PAF, and MCI-LB, but in principle also some less specific syndromes and symptoms, such as hyposmia, constipation, depression, and anxiety. Finally, there is advanced disease.
The preclinical stage is characterized by formation and initial propagation of aggregated αSyn species. By analyzing data from two postmortem cohorts, we have previously shown that the majority of brains with very restricted incidental Lewy body disease can be divided into two major categories [85, 90]: (i) a brain-first group with Lewy pathology in the olfactory bulb, limbic system and top of the brainstem, but without pathology in the lower brainstem, spinal cord, or peripheral autonomic nervous system, and (ii) a body-first group with Lewy pathology in the peripheral autonomic nervous system, lower brainstem, and spinal cord, but without pathology in the olfactory bulb, limbic system, or upper brainstem. Additionally, a small mixed group can be defined, exhibiting pathology in olfactory/limbic structures, as well as in autonomic and lower brainstem structures. This group, however, lacks pathology in the intervening structures, including the upper brainstem. These mixed cases were relatively rare in our analysis (<10%), and thus the majority of cases are compatible with the pathology having started in a single, restricted region, either the olfactory bulb or enteric nervous system, with secondary spreading from those two originating sites [85, 110].
Pure autonomic failure (PAF) is a rare clinical diagnosis characterized by predominantly peripheral autonomic symptoms with few or no CNS symptoms [111]. Patients universally show severe autonomic denervation on MIBG scintigraphy, but their dopamine and other CNS systems are typically much more normal [112, 113]. Only few PAF cases have come to autopsy, but these typically showed Lewy pathology restricted to the lower brainstem or in some cases higher Braak stages [114–119]. At follow-up, many PAF patients convert to PD or DLB, and those who do are almost always RBD-positive [112, 120]. This points to a relatively stereotypical trajectory from PAF to iRBD and finally to PD or DLB. Of note, PAF is rarely diagnosed. The majority of individuals with iRBD are not typically diagnosed with PAF before receiving an iRBD diagnosis, although a large percentage of iRBD patients do exhibit orthostatic hypotension, which in some cases may have preceded the emergence of RBD itself [121]. Such iRBD patients could therefore have been eligible for the PAF diagnosis.
Patients with Lewy body-positive iRBD convert to PD (54%) or DLB (46%) [122]. Although postmortem data on iRBD cases are rare, the published cases showed Lewy pathology in the brain, typically Braak stages 3-5 [123–126]. In vivo, most iRBD cases show Lewy pathology in peripheral autonomic nervous fibers, i.e., in salivary glands, skin, and colon although limited reproducibility has been reported for colon biopsies [127–130]. Patients with iRBD exhibit almost universal loss of the cardiac sympathetic system on MIBG imaging [131–133], decreased cholinergic innervation of the colon signifying parasympathetic denervation [131], low neuromelanin signal of the locus coeruleus [131, 134], but commonly show normal or near-normal dopamine transporter imaging [131, 132]. Thus, patients with iRBD show neuronal degeneration in the lower brainstem and autonomic system and progresses to either PD or DLB in almost equal proportions.
According to the Brain- vs. Body-first model, an olfactory-first or limbic-first type of Lewy body disease leads to clinical brain-first PD subtype, characterized by fairly asymmetric dopamine degeneration [78]. At diagnosis, such patients have fewer non-motor symptoms and are typically RBD-negative [52, 84]. On imaging, these de novo brain-first PD patients show limited or no damage to the locus coeruleus, parasympathetic, or sympathetic systems [52, 136]. However, on follow-up most of these initially RBD-negative PD patients will eventually develop RBD and an increasing burden of non-motor symptoms. In support, it has been shown that 87% of late-stage PD and more than 75% of DLB patients are RBD-positive, whereas only about 1/3 of PD patients developed RBD before diagnosis [55, 138].
Although follow-up studies of well-characterized brain-first PD patients are lacking, it is known that virtually all PD patients at H&Y stage III show severe cardiac denervation on MIBG scintigraphy [53, 54]. Thus, it seems likely that most brain-first patients will eventually converge on the same advanced Lewy body stage, characterized by neurodegeneration across all vulnerable peripheral and CNS nuclei and accompanied by RBD and clinical dementia. In support, studies have estimated that at least 80% of all PD patients develop dementia during life [72, 140], and it remains possible that indeed all patients with clinical Lewy body disease will eventually dement, if they survive long enough.
For completeness, Fig. 3 includes a clinical category termed “non-PD/non-RBD”. Among patients in the advanced Lewy body dementia stage, the majority had developed parkinsonism, RBD, or both prior to onset of dementia. Such patients are captured by the PD and iRBD categories in the framework, but that still leaves a small group of pre-demented symptomatic patients without parkinsonism or RBD. Plausibly, such patients might have hyposmia, neuropsychiatric symptoms including anxiety, depression, and hallucinations, and MCI [57]. It is possible that mixed AD-Lewy body pathology could be particularly common in this patient group, leading to accelerated psychiatric symptoms and cognitive decline due to significant co-morbid AD pathology. The proposed ‘psychiatric-onset’ form of DLB, for which there is currently insufficient evidence to support distinct clinical criteria, could be more prevalent in this group, but it is unknown whether this type of DLB exhibits higher loads of AD co-pathology [141]. In any case, some of these patients reach the advanced dementia stage before parkinsonism or RBD has appeared, but these symptoms are likely to appear during the dementia stage. In support, it has been shown that a small percentage (10–15%) of DLB patients show normal dopamine imaging at diagnosis, but these will eventually develop dopamine degeneration upon follow-up [48]. It is also important to note that some individuals in the “non-PD / non-RBD” category may develop parkinsonism before dementia and thus fulfil the criteria for PD before they reach the advanced stage.
LIMITATIONS
The framework suggested here (Fig. 3) is based on integrating the brain- vs. body-first model with other concepts and observations from the literature. This model is still hypothetical, and its limitations have been thoroughly reviewed previously [52, 110]. While the model proposes that the specific origin site of initial Lewy pathology and its subsequent propagation through the connectome is a major determining factor for the evolution of different clinical phenotypes, we want to emphasize that many other pathogenic factors, such as genetic variants, inflammation, and selective neuronal vulnerability factors, are important for the shaping the variable disease course in individual patients.
Our proposed framework is meant to explain the relationship between progressive Lewy pathology, co-pathologies, and different clinical syndromes. It is not intended as a staging or diagnostic system in its current form. Several obstacles remain with regards to the validity of both this framework and recently published biological staging systems [8, 9], such as determining the sensitivity and specificity of synuclein seed amplification assays to diagnose prodromal and preclinical Lewy body disease [130, 143].
More work is certainly needed with respect to understanding different clinical phenotypes and their underlying pathophysiological causes, including PAF and psychiatric-onset DLB. Such distinct clinical phenotypes may be caused by particular individual vulnerability factors interacting with pathological α-synuclein and other aggregated protein pathologies.
Finally, we must always consider carefully how changes in diagnostic criteria or syndromic conceptualization may adversely impact patient care and advocacy efforts.
SUMMARY
In the framework presented here, we propose that the different clinical syndromes, such as iRBD, PAF, PD, PDD, and DLB, should be viewed as manifestations of a common underlying Lewy body disease, but which can be modified by genetic variants, selective neuronal vulnerability factors, and the presence of various co-pathologies, with the most significant being AD pathology.
Patients in the earliest clinical disease stages will show the greatest degree of heterogeneity, since the pathogenic process can originate in different locations of the neuroaxis and the associated neurodegeneration is still relatively localized. However, the majority of patients with Lewy body disease appears to progress towards a more homogenous advanced disease stage. In this stage, currently diagnosed as PDD or DLB, most patients will exhibit a full-spectrum clinical phenotype, including dementia, psychiatric manifestations, parkinsonism, RBD, autonomic issues, and other non-motor symptoms. The imaging literature also supports that patients with advanced disease commonly show evidence of neurodegeneration across all major neurotransmitter systems known to be vulnerable to Lewy pathology, which is not the case for early-stage disease.
Future research should focus on understanding how different clinical signs and symptoms map to underlying neuronal dysfunction and damage, including brain region-specific changes, with the goal of facilitating personalized treatments. This may potentially lead to biological subtypes of Lewy body disease, if it can be demonstrated that they have a specific biological-clinical “fingerprint”. But the best starting point is to acknowledge that this disease does not conform to our current clinical diagnostic criteria and can manifest itself quite variably, in terms of age of onset, organ and neurotransmitter systems affected, symptom presentation and evolution over time, and disease progression over many years.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflicts of interest to report.
Daniel Weintraub is an Editorial Board Member of this journal but was not involved in the peer-review process nor had access to any information regarding its peer-review.