
Abstract
The protein alpha-Synuclein (α-Syn) is a key contributor to the etiology of Parkinson’s disease (PD) with aggregation, trans-neuronal spread, and/or depletion of α-Syn being viewed as crucial events in the molecular processes that result in neurodegeneration. The exact succession of pathological occurrences that lead to neuronal death are still largely unknown and are likely to be multifactorial in nature. Despite this unknown, α-Syn dose and stability, autophagy-lysosomal dysfunction, and inflammation, amongst other cellular impairments, have all been described as participatory events in the neurodegenerative process. To that end, in this review we discuss the logical points for gene therapy to intervene in α-Syn-mediated disease and review the preclinical body of work where gene therapy has been used, or could conceptually be used, to ameliorate α-Syn induced neurotoxicity. We discuss gene therapy in the traditional sense of modulating gene expression, as well as the use of viral vectors and nanoparticles as methods to deliver other therapeutic modalities.
BACKGROUND
Parkinson’s disease (PD) is a neurodegenerative disorder clinically characterized by cardinal motor symptoms which can be attributed to the loss of striatal dopaminergic tone and subsequent loss of dopaminergic neurons in the substantia nigra. Postmortem evaluation of PD patient brains has revealed the presence of proteinaceous cytosolic inclusions, termed Lewy bodies (LB), and thread-like fibrils in cellular processes, termed Lewy neurites (LN), in neurons throughout various brain regions. Genetic studies linked the protein alpha-Synuclein (α-Syn) to familial forms of the disease and subsequent studies identified α-Syn as a major component of LBs [1]. Moreover, point mutations and gene multiplications of SNCA alter the aggregation potential of α-Syn and cause PD in a dose-dependent manner [2, 3], thus, indisputably linking α-Syn to the disease process.
The exact function(s) of α-Syn is still largely unknown. α-Syn, located in synaptic terminals and neuronal nuclei in the central and peripheral nervous systems is typically viewed as a neuronal protein involved in neurotransmission. However, the protein is also expressed in a variety of non-neuronal tissues including cells from a hematopoietic origin [4], suggesting a function that extends beyond the nervous system. Under normal conditions α-Syn is a natively unfolded and soluble monomer, with its existence as a tetramer debated [5, 6]. However, during the process of pathogenesis, α-Syn misfolds and forms aggregates along with other proteins, forming LN and LB, collectively referred to as Lewy pathology (LP). Aberrant neuronal accumulation of α-Syn has also been identified in dementia with Lewy bodies (DLB), and oligodendroglial accumulation of α-Syn is seen in multiple system atrophy (MSA) (reviewed further in [7, 8]). How the same protein is able to cause different diseases in different cell types still remains unclear; however, the link of α-Syn aggregation has collectively united these diseases under the umbrella term synucleinopathies and highlights α-Syn as a central therapeutic target [9].
α-SYNUCLEIN IN PD PATHOLOGY
By comparing the brains from patients presenting a spectrum of PD-associated symptoms and LP, Braak and colleagues conceived a model for the etiology of PD, in which the LP begins in the brainstem and/or olfactory bulb and extends rostral or caudally to other brain regions over time [10]. This model led to the creation of the Braak hypothesis, which in its latest iteration proposes that α-Syn pathology of PD starts in the gut (possibly initiated by an unknown pathogen) or the olfactory bulb and spreads towards, and within, the CNS in a prescribed temporal and spatial distribution. As pathology progresses, neuronal dysfunction, possibly related to α-Syn pathology per se, gives result to prodromal symptoms, and eventually classical parkinsonism ensues following the involvement of the basal ganglia. This idea has given rise to several lines of research: 1) PD prodrome; or the presence of premotor disease (i.e., Braak Stages 1 and 2); 2) PD co-pathology; the extent to which α-Syn pathology is informed by pathologies other than LP (e.g., tau [11]), and 3) PD as a prion-like disorder. Nonetheless, one commonality that ties these areas of research together is the presence of α-Syn pathology in one form or another.
RATIONALE FOR GENE THERAPY-MEDIATED TARGETING α-SYN IN DISEASE: TOXIC GAIN-OF-FUNCTION
A prion-like misfolding [12] and propagation of α-Syn non-monomeric species that eventually become insoluble aggregates is proposed as an event necessary for neurodegeneration [13]. The idea of active propagation has been supported by studies showing that injections of α-Syn fibrils can recruit adjacent neurons along the olfactory tract [14] or vagus nerve [15, 16]. With propagation proposed to occur through exosomes or via neuronal release and endocytosis, directly between neurons, as well as through the involvement of microglia [17]. However, the results as it relates to the propagation of α-Syn from the periphery per se have been mixed [18–20], with some studies reporting absent or transient pathology, and most studies demonstrating sustained CNS pathology following a peripheral inoculation requiring an additional “hit” such as a mutant α-syn transgenic background [21].To this end, it is important to note the limitations of these animal models, as they do not reflect “normal” PD pathophysiology in the sense that an injection of supraphysiological concentrations of α-Syn is required [17].
Although the exact role of α-Syn in disease remains largely unknown, the chief presumption is that α-Syn aggregation causes neurodegeneration via a direct toxic gain-of function. Recent work has highlighted that not fibrils per se, but the entire process of LB formation-including fibrilization, posttranslational modifications, and interaction with membranous organelles is the key driver of toxicity, by disrupting essential cellular functions and inducing synaptic dysfunction, as well as mitochondrial toxicity [22]. As such, a variety of therapeutic modalities have been conceived to target α-Syn protein levels and aggregate formation.
GENE THERAPY INTERVENTIONS BASED ON α-SYN
Based on the assumption that α-Syn pathology is a cause and not a consequence of disease, anti-Synuclein strategies have emerged as the indisputable disease-modifying therapeutic strategy, akin to a similar framework of anti-amyloid interventions in Alzheimer’s disease. The overarching goal aims to reduce the load of α-Syn pathology through a variety of means-either by targeting the process of aggregation or by targeting the consequences of aggregation. Thus far, the only clinical anti-Synuclein strategies have utilized active or passive immunization [23, 24]. Although available results to date have failed to meet the primary objective, the analysis of secondary outcome measures have signaled improvement. Nonetheless, numerous modalities aimed at utilizing gene therapy to target α-Syn pathology are in various stages of preclinical investigation.
In the strictest sense, gene therapy is defined as technique that modifies an individual’s genetic makeup to treat or cure disease. Below we discuss α-Syn gene therapy strategies based on existing preclinical gene therapy studies, as well as considering conceptual strategies based on basic research into α-Syn biology.
As depicted in Fig. 1, there are several logical points whereby one could utilize gene therapy to target α-Syn pathology: 1) Target extracellular α-Syn (the presumptive prion pathogen) using immunotherapy; 2) Blocking or reducing expression of receptors that may facilitate cell-to-cell propagation; 3) Use RNAi or similar technologies to decrease overall levels of α-Syn; 4) Utilize strategies that stabilize the monomeric (functional) form of the protein or enhance clearance of aggregated protein; 5) Promote cellular processes that are impaired due to α-Syn aggregation; 6) Target inflammation; 7) A toxic loss-of function hypothesis will be discussed in detail below, but with this idea in mind, one therapeutic approach may be to maintain monomeric forms of the protein.
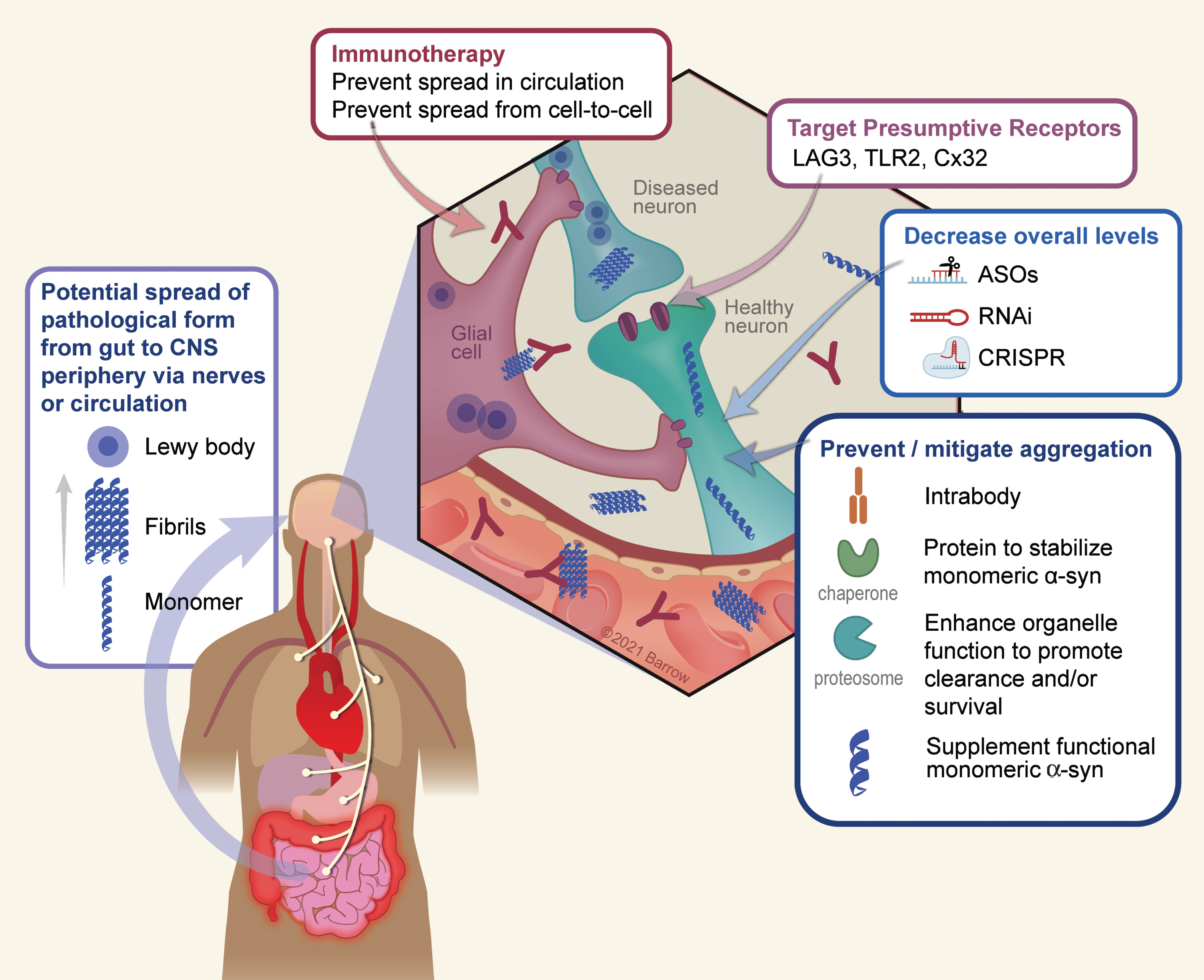
Conceptual points of intervention in α-Syn gene therapy. There are numerous conceptual points of gene therapy intervention aimed at preventing or ameliorating toxic effects that arise as a result of α-Syn oligomerization or depletion in disease. 1) A growing body of data suggests a peripheral origin of Lewy pathology which spreads rostrally to the CNS, and thereafter throughout the PD brain. Although the exact mechanism by which this occurs is still unknown, it is thought to involve extracellular α-Syn which would serve as a substrate for immunotherapy. 2) The surface proteins LAG3, TLR2, and Cx32 can interact with α-Syn and may mediate neuronal uptake of pathological forms of the protein. Accordingly, targeting these receptors either via immunotherapy or genetically (e.g., via RNAi) is a potential means to prevent trans-neuronal spread of pathology. 3) A chief strategy thus far has been to utilize various genetic means such as anti-sense oligonucleotides, RNA interference, or CRISPR-based technology to lower the overall dose of the protein and thus reducing the ability of α-Syn to aggregate. 4) A second approach to reduce aggregation is to directly stabilize the monomeric, soluble, form of α-Syn using chaperones or intrabodies. Along the same lines, enhancing the clearance of intracellular α-Syn aggregates via the enhancement of autophagy/lysosomal function, can also serve as to minimize the degree of aggregation. 5) Finally, in the process of aggregation the soluble pool of α-Syn is depleted, resulting in a potentially toxic loss-of function. Supplementation of non-aggregatable forms of the protein can then be introduced to restore crucial protein function. Used with permission from Barrow Neurological Institute, Phoenix, Arizona.
GENE THERAPY TO PREVENT α-SYN PATHOLOGY PROPAGATION
As mentioned above, the main approach to target extracellular α-Syn, and thus cell-to-cell spread, has taken place via classic immunotherapy [23]. However, vectorized immunotherapy, in which antibodies are directly delivered to the CNS using viral vectors, is being explored in attempts to enhance target engagement within the parenchyma. This approach is under development by Voyager Therapeutics, although the status of these studies is unknown. Nonetheless, with the advent and success of mRNA vaccines in the era of COVID-19, similar immune-based gene therapy approaches targeting α-Syn are likely to follow. Moreover, the use of gene therapy to directly produce intrabodies within target cells holds significant promise in reducing α-Syn related pathology [25, 26].
The exact cell surface receptor(s) involved in fibril internalization are unknown, but some potential candidates for anti-propagation therapies have been proposed: Connexin-32 (Cx32) [27], Toll-Like receptor 2 (TLR2) [28], and Lymphocyte-activation gene 3 (LAG3) [29], although the relevance of the latter in neurodegeneration is debated [30]. These proteins represent viable gene therapy targets by either RNA interference (RNAi) or CRISPR-based approaches to ameliorate symptoms of neurodegeneration.
GENE THERAPY TO REDUCE EXPRESSION OF α-SYN
Targeted gene silencing of α-Syn has been performed with various approaches including antisense oligonucleotides (ASOs), short interfering RNA (siRNAs), short hairpin RNA (shRNA) and zinc finger nucleases, with both liposomal and viral vectors as the delivery modality [31–34]. More recently, CRISPR-based technologies have also been utilized to modulate α-Syn expression via transcriptional regulation through the endonuclease deficient dCas9-based system [35]. While, many studies suggest α-Syn silencing may prove beneficial [31, 36–38] in preventing α-Syn toxicity a number of studies from our group, in rodent and non-human primates, show nigrostriatal degeneration as a direct result of α-Syn knockdown [32, 40]. The source of this discrepancy is unknown but may be attributed towards compensatory increases in β-Syn and γ-Syn levels, which may share a common redundant function with α-Syn [41]. The compensatory effect of these proteins may be facilitated by differences in the kinetics and duration of α-Syn suppression (e.g., contrasting the work by Benskey [32] with that of Zharikov [36], the former describing a much more rapid removal of nigrostriatal α-Syn and nigrostriatal degeneration). Thus, the strategy of reducing the soluble form of α-Syn should carefully consider the potential toxic effects of an excessive reduction of the physiological levels of α-Syn below a certain threshold necessary for normal function (see more below).
GENE THERAPY STRATEGIES TO STABILIZE THE MONOMERIC (FUNCTIONAL) FORM OF THE PROTEIN OR ENHANCE CLEARANCE OF AGGREGATED PROTEIN
The stabilization of monomeric species of α-Syn, or the breakdown of fibrils are possible strategies to reduce α-Syn aggregation, ultimately protecting the soluble pool of this peptide. This has been achieved by different molecules, such as catecholamines, natural phenols, or synthetic compounds [42–44]. In essence, this approach exploits the intrinsic biochemical characteristics and the complex structure of α-Syn. These include binding to the negatively charged C-terminus domain (e.g., catecholamines), or interference with the intramolecular long-range interaction between N- and C-terminus by N-terminus residues (e.g., CLR01), or binding to the hydrophobic sites of the oligomeric species (e.g., Anle138b) [45–48]. A direct effect of stabilizing the monomeric form of the protein provides an environment that thermodynamically favors soluble α-Syn and leads to the amelioration or prevention of the α-Syn nucleation process [43, 44].
Although small molecules have been in the forefront of stabilizing monomeric α-Syn, several gene therapy candidates, which may serve analogous functions, have emerged. Intrabodies (nanobodies) engineered against the non-amyloidogenic portion (NAC) of α-Syn can both inhibit misfolding, as well as enhance the clearance of the protein, reducing toxicity both in in vitro and in vivo synucleinopathy models [25, 26]. Similarly, overexpression of chaperones such as HSP-70 can ameliorate α-Syn-mediated toxicity, presumably by preventing fibril formation [49–51].
Impairments in the autophagy-lysosomal pathway (ALP) have been linked to PD. The most common genetic risk factors for PD are Gaucher disease variants (GBA), which cause the loss of function of the lysosomal glucocerebrosidase enzyme (GCase) [52]. In vitro, the accumulated GCase substrate, glycosylceramide, acts like a scaffold promoting the aggregation of α-Syn oligomers and fibrils [53]. Conversely, α-Syn pathology can inhibit GCase activity [54], thus, suggesting a feed-forward loop linking α-Syn and GCase [55]. To this end, numerous gene therapy-based approaches have been employed to improve ALP function. Overexpression of GCase itself can reduce α-Syn aggregation and toxicity [56, 57]. Similarly, overexpression of proteins aimed at strengthening ALP such as: Beclin-1 [58], the transcription factor EB (TFEB; a key regulator of ALP) [59], and LAMP2a [60] reduces nigral toxicity in various models of α-Syn pathology. Moreover, the microRNA miR-124 regulates numerous genes involved in ALP, and its expression is deregulated in PD. Accordingly, ectopic modulation of this microRNA is viewed as a potential modality in targeting α-Syn pathology [61, 62].
GENE THERAPY FOR UNIFORMITY ALLEVIATE INFLAMMATION
Several reports implicate the immune system in the pathophysiology of sporadic PD [63, 64]. Microglial activation [65, 66], T-cell activation [67], and increased inflammatory cytokine production [68], are documented in sporadic PD patients, as well as in MSA animal models and MSA patients [69]. Several factors including genetics, and infectious agents can trigger an immune-mediated inflammatory response. Activated microglia can stimulate T-cells to produce and directly release inflammatory cytokines including L-1β, IL-2, IL-6, EGF, and TGF-α and TGF-β [67]. The results of such inflammatory processes are now thought to directly contribute towards neurodegeneration. Moreover, the neurodegenerative process per se could activate microglia and further exacerbate neuronal death [66]. Once the degenerative neuroinflammatory process starts, the blood-brain barrier is weakened, becoming permeable to peripheral immune cells. This amplifies the inflammatory response and facilitates the transition toward a chronic inflammatory state. The large body of work that is immunology confers numerous potential gene therapy targets: One can envision to manipulate proteins that are 1) directly involved in α-Syn-mediated inflammatory processes, or 2) those that are general participants in inflammation. For example, overexpression of fractalkine [70] or a dominant negative form of TNF [71] attenuates microglial activation, and protects against α-Syn overexpression or treatment with the parkinsonian 6-OHDA toxin, respectively. Despite a conceivably long list of potential genetic targets in neuroinflammation, few studies have used gene therapy to manipulate these targets. One reason may reside in the difficulty in targeting immune cells in the brains using conventional vectors. For example, microglial cells remain relatively refractory to infection with common viral vectors. Furthermore, it is becoming increasingly appreciated that subpopulations of glia exist within diseased tissue, exhibiting divergent roles (e.g., neuroprotective versus neurotoxic) in the disease process. Targeting such glial subpopulations will thus require a new generation of viral vectors with much improved precision and efficacy.
SUPPLEMENTING SOLUBLE α-SYN TO RETAIN FUNCTION: THE LOSS-OF FUNCTION HYPOTHESIS
The chief preclinical focus in PD is directed towards the direct toxic effect of α-Syn aggregation, while very little attention has been given to potential toxic effects resulting from α-Syn depletion due to aggregation; namely the loss-of-function (LOF) hypothesis. As described above, multiple studies have demonstrated toxic effects due to rapid α-Syn removal in PD-susceptible populations of neurons. Moreover, several lines of evidence show that monomeric soluble α-Syn is depleted during the process of aggregation resulting in a de facto physical LOF [72, 73], where the monomers are sequestered in a non-native conformation within solid amyloid fibrils. Despite the difference in mechanism, both genetic and physical LOF will confer the same pathophysiological consequences as a result of α-Syn depletion.
Despite the lack of universal agreement on its precise function, a wealth of studies implicates a role for α-Syn in a variety of essential processes, including synaptic vesicle trafficking and neurotransmitter release [74], immune cell maturation and function [4, 76], DNA repair [77], and dopamine biosynthesis [78]. Thus, it is not inconceivable that a protein with crucial function(s) and with an abundant expression pattern throughout the body, when perturbed, can have deleterious consequences to the cell, as well as the organism as a whole.
Several pathological factors can push a protein over the physical nucleation barrier via different pathways and trigger amyloid aggregation. Protein overexpression due to gene duplication and triplication can lower the nucleation barrier increasing the probability of spontaneous nucleation. Furthermore, adding preformed nuclei (seeds/prions) will catalyze amyloid transformation by bypassing the nucleation step altogether. Moreover, exogenous surfaces such as nanoparticles or microbes can catalyze nucleation by increasing local protein concentration and inducing conformational changes via heterogeneous nucleation [79]. Many proteins exposed to such catalytic pathways of nucleation will end up in the non-functional amyloid state, while not necessarily becoming more toxic in the process. Thus, although controversial, LOF needs to be considered as a potential pathogenic mechanism in PD. To that end, strategies that aim to stabilize monomeric forms of the protein, or to remove the nucleation seed (i.e., aggregated Synuclein) are ideal therapeutic candidates. Moreover, supplementation in the form of non-aggregatable forms of the protein should also be considered.
CONCLUSIONS
Without doubt, α-Syn is a key etiopathological participant in PD and other synucleinopathies, and at the center of the role of α-Syn in disease is the propensity of this protein to form stable aggregates. The structure and formation of such α-Syn aggregates confers detrimental effects on neurons and depending on the vulnerability of specific neuronal sub-population neurodegeneration occurs. Nonetheless, the specific mechanism by which the neuron is impacted, whether it is toxic gain or loss of function, and whether α-Syn-mediated toxicity is heterogenous in nature remains elusive. Still, in the last decade, numerous preclinical gene-therapy targets have emerged, representing conceptually distinct potential points of intervention as it relates to various molecular processes in neurons. At the same time, CNS gene therapy has made great strides, and PD has a rich history utilizing gene therapy, with 25 trials currently listed on clinicaltrials.gov involving PD, albeit none have been aimed at modulating α-Syn. Current and completed studies have focused on the neuroprotective effects of neurotrophic factors glial cell line-derived neurotrophic factor (GDNF, NCT01621581) and neurturin (CERE-120) [80], as well as to increase enzymatic levels such as glutamic acid decarboxylase (GAD) [81], aromatic L-amino acid decarboxylase (AADC; NCT03065192), or a combination of enzymes with lentiviral delivery of tyrosine hydroxylase, AADC, and GTP cyclohydrolase 1 (Prosavin; [82]). While clinical trials have yielded varying degrees of clinical improvements in PD motor symptoms, gene therapy studies have proven long-term safety [83]. With the concomitant development of novel, more efficient means of delivery, we are likely to see α-Syn-specific gene therapies in the years to come.
Footnotes
ACKNOWLEDGMENTS
Authors are supported by the Barrow Neurological Foundation.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.