
Abstract
Background:
Parkinson’s disease (PD) is a progressive neurodegenerative disorder thought to be caused by accumulation of α-synuclein (α-syn) within the brain, autonomic nerves, and the enteric nervous system (ENS). Involvement of the ENS in PD often precedes the onset of the classic motor signs of PD by many years at a time when severe constipation represents a major morbidity. Studies conducted in vitro and in vivo, have shown that squalamine, a zwitterionic amphipathic aminosterol, originally isolated from the liver of the dogfish shark, effectively displaces membrane-bound α-syn.
Objective:
Here we explore the electrophysiological effect of squalamine on the gastrointestinal (GI) tract of mouse models of PD engineered to express the highly aggregating A53T human α-syn mutant.
Methods:
GI motility and in vivo response to oral squalamine in PD model mice and controls were assessed using an in vitro tissue motility protocol and via fecal pellet output. Vagal afferent response to squalamine was measured using extracellular mesenteric nerve recordings from the jejunum. Whole cell patch clamp was performed to measure response to squalamine in the myenteric plexus.
Results:
Squalamine effectively restores disordered colonic motility in vivo and within minutes of local application to the bowel. We show that topical squalamine exposure to intrinsic primary afferent neurons (IPANs) of the ENS rapidly restores excitability.
Conclusion:
These observations may help to explain how squalamine may promote gut propulsive activity through local effects on IPANs in the ENS, and further support its possible utility in the treatment of constipation in patients with PD.
Keywords
INTRODUCTION
Parkinson’s disease (PD) is a progressive neurodegenerative disease associated with the accumulation of the protein α-synuclein (α-syn) within the peripheral and central nervous systems (CNS) [1]. Diagnosis of PD is primarily based on the presence of a combination of motor and non-motor symptoms [2]. The latter include severe neuropathic constipation which presents an important therapeutic challenge [3]. Lewy bodies containing α-syn are a pathognomonic sign of PD and are classically found in the CNS. However, they can also be detected within the enteric nervous system (ENS) of patients with sporadic disease [4, 5] and have been detected in the ENS before the clinical diagnosis was made [6]. In a majority of people with PD, constipation occurs many years before the onset of motor symptoms [7–9].
According to Braak’s hypothesis PD may begin peripherally with the development of misfolded α-syn aggregates (Lewy bodies) in the olfactory nerves, submandibular gland and ENS [4, 5], then traffic to the CNS. In the case of the gut they move by vagal preganglionic fibres that arise from viscero-motor projection neurons of the dorsal motor nucleus (DMV) [1]. This hypothesis has been supported recently by experiments in rodents. α-syn from PD patient brain lysate injected into the intestine progressed to the brainstem [10]. α-syn preformed fibrils injected into the duodenal and pyloric muscularis layer were observed in the DMV and then dispersed caudally in the hindbrain, eventually reaching the basolateral amygdala, dorsal raphe nucleus, and substantia nigra pars compacta [11]. Truncal vagotomy prevented gut to brain spread of α-syn pathology [11]. In another study, α-syn preformed fibrils, injected into the duodenal wall of bacterial artificial chromosome (BAC) transgenic rats, propagated to the DMV as well as via a sympathetic route to the celiac ganglia and the intermediolateral nucleus of the spinal cord (IML) [12]. Additionally, low-dose, chronic administration of the pesticide rotenone in mice induced α-syn aggregation in the ENS, DMV, intermediolateral nucleus of the spinal cord, and the substantia nigra, reproducing PD pathological staging [13]. This progression was preventable by vagotomy [14]. Clinically Lewy bodies have been detected in the vagus of patients with PD [15] and a single large-scale retrospective epidemiological study of vagotomy in humans showed a reduced risk of development of PD in those patients who received truncal vagotomy [16]. However, some evidence suggests that ENS spread of α-syn pathology to the CNS after exposure may not be sufficiently sustained and may require other factors [17], although this may be influenced by differing experimental methodologies including type and quantity of α-syn and location of injection [11].
Ninety to 95% of the sensory neurites that innervate the intestinal epithelium originate from the ENS, with the rest arising from cell bodies outside the intestine [18, 19]. Intrinsic primary afferent neurons (IPANs) are ENS sensory neurons in the myenteric plexus that are essential for normal gut propulsive peristalsis and indeed life itself as their selective silencing in transgenic mice leads to death shortly after weaning [20]. We have shown recently that two thirds or more of intestinal vagal afferent signaling is relayed via IPANs through an IPANs-to-vagus intramural sensory synapse [21]. If myenteric IPANs’ excitability is compromised in the hSNCAA53T model used in the present report, constitutive afferent vagal firing rates could be reduced due to decreased activity at the intramural sensory synapse. To test this, we have measured vagal afferent firing rates from the mesenteric nerve bundle of the small intestine of the hSNCAA53T model mice and controls. Furthermore, we have examined these rates before and after adding squalamine to the gut lumen.
Squalamine is a natural zwitterionic amphipathic aminosterol with a net positive charge [22], produced in the liver of the dogfish shark and secreted along with bile salts into the proximal duodenum [23]. The synthetic squalamine salt (ENT-01), inhibits the formation of neurotoxic α-syn aggregates in a C. elegans model of PD, both in vitro, and in vivo [24]. We have recently demonstrated that acute intraluminal administration of squalamine reverses the reduced colonic propulsive motility and decreased constitutive vagus nerve firing observed in aged (≥2 y) compared to young (3 month) male mice [25]. The clinical prokinetic effect in gut motility appears to occur through local action of the compound on the ENS, since squalamine is not significantly absorbed into the systemic circulation [26].
In the present studies we explore the electrophysiologic effect of squalamine on the gastrointestinal (GI) tract of both normal mice and two mouse models of PD that express the highly aggregating A53T mutant form of human α-syn. The first model used was a mouse α-syn knock-out/knock in model that expressed four copies of human A53T α-syn (hSNCAA53T) driven by the endogenous α-syn promoter but lacking the murine α-syn coding sequence (mSnca). The control is represented by a strain engineered to express two copies of the normal human α-syn protein (hSNCA) but also lacking the endogenous mouse α-syn coding region (mSnca) [27]. This model (dbl-PAC-Tg (SNCAA53T), will be referred to as hSNCAA53T in the subsequent text and in corresponding figures. The hSNCAA53T strain exhibits a constipation phenotype that is more severe than that observed for the corresponding control strain [27]. These animals represent an early model of familial PD, as they display ENS dysfunction by 3 months, progressing in severity to 6 months, in the absence of CNS pathologies such as olfactory dysfunction, dopaminergic deficits, Lewy body inclusions, and neurodegeneration [27, 28]. A recent study by the same group demonstrated that the translational inhibitor of α-syn, Posiphen, normalized colonic dysmotility in the hSNCAA53T strain [28].
The second model, homozygotic A53T human α-syn overexpressing mice (7 months) [29] and their non-transgenic (non-Tg/WT) littermate controls (7 months), were compared to assess the effect of α-syn aggregation on colonic transit in another PD model. In this engineered mouse model, human A53T (hSNCAA53T) expression is driven by a mouse prion promoter (PrP) resulting in the progressive accumulation of aggregates of A53T α-syn throughout the nervous system [29]. These mice are referred to as PrP-A53T and their controls as non-Tg in the following text and figures. When the homozygotes (PrP-A53T mice) reach an age of 7–8 months they begin to develop progressive impairment of motor function so severe that they are eventually unable to support themselves to feed and succumb at about 16 months [29]. Unlike the hSNCAA53T model, PrP-driven human A53T α-syn expression results in accumulation of filamentous α-syn aggregates in the CNS leading to late-onset neurodegeneration (>6 months) and mimicking human familial PD neuropathology [29].
We show here that colonic motility is impaired in both mouse models of PD, accompanied by reduced firing rate in vagal nerve fibres of the mesenteric nerve bundle and electrical behavior of IPANs in the hSNCAA53T model. Application of squalamine rapidly restores the normal firing rate and electrical behavior together with promotion of propulsive activity.
MATERIALS AND METHODS
All animal studies were approved by and performed in accordance with the Animal Research Ethics Board (AREB) of McMaster University (permit 16-08-30) and of the Florey Institute of Neuroscience and Mental Health (approval 16-029).
hSNCAA53T mice and their controls (endogenous promotor) used for motility (Fig. 1), mesenteric nerve recording (Fig. 3), and electrophysiological recordings (Figs. 4 and 5) were obtained from Jackson Laboratories (Maine, USA). 13–16 male PAC-Tg (SNCAWT) (Stock No. 010710; Control) and (dbl-PAC-Tg (SNCAA53T) (Stock No. 010799; PD mice) were aged 8–9 months prior to experiments. Researchers were blinded to animal groups.
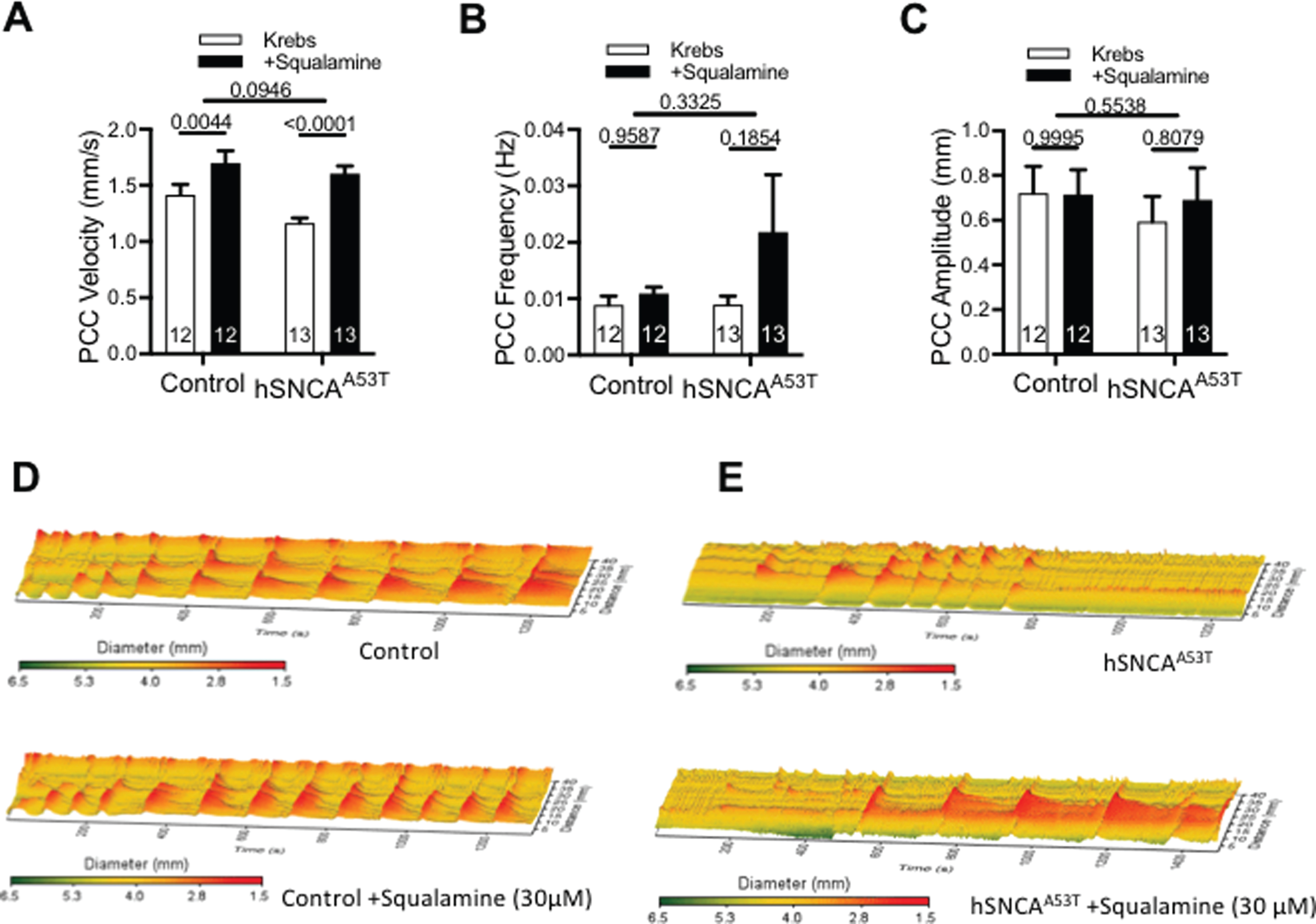
Colonic motor activity was increased with intraluminal squalamine in both hSNCAA53T (endogenous promoter) mice and control mice. A) hSNCAA53T mice did not have reduced PCC velocity compared to control mice (p = 0.0946). Intraluminal squalamine (30μM) significantly increased colonic PCC velocity in control and hSNCAA53T mice (N = 12–13 mice/group, 2-way ANOVA with Sidak’s multiple comparisons test). B) hSNCAA53T mice did not have reduced colonic frequency compared to control, nor did squalamine produce an effect on colonic frequency (N = 12–13 mice/group, 2-way ANOVA with Sidak’s multiple comparisons test). C) hSNCAA53T mice did not have reduced colonic amplitude compared to control, nor did squalamine produce an effect on colonic amplitude (N = 12–13 mice/group, 2-way ANOVA with Sidak’s multiple comparisons test). D) Control mice had more uniform and consistent colonic contractions during Krebs control compared to (E) hSNCAA53T mice who had infrequent shorter contractions as represented in the 3D heat maps. Intraluminal squalamine increased colonic contractions, particularly in hSNCAA53T mice. Contractions (red) and relaxation (yellow-green) are depicted across time and distance along the colon. All data represented as mean±S.E.M, *p < 0.05.
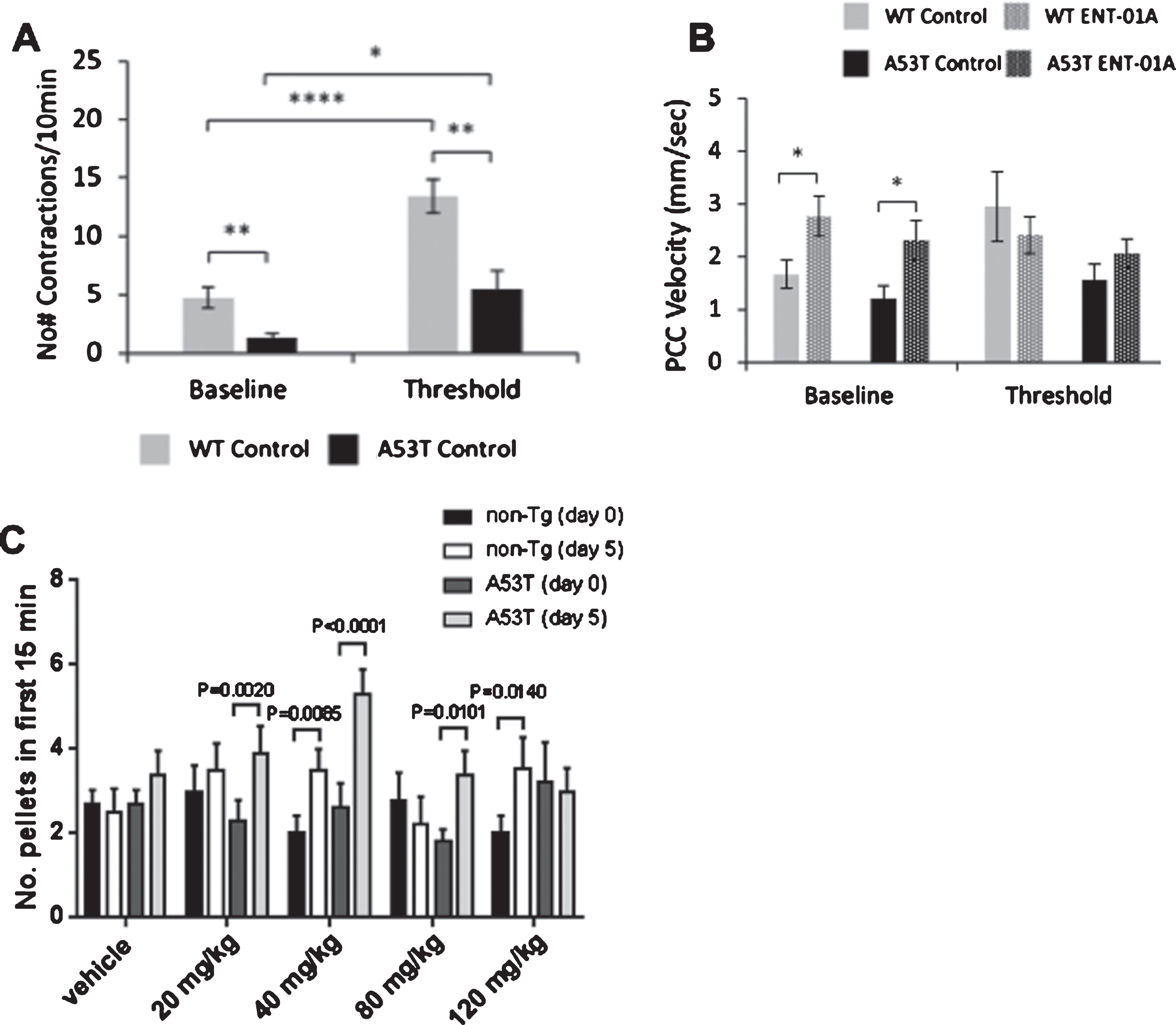
A53T (prion promoter) mice had reduced colonic motor activity compared to non-Tg control mice but improved fecal pellet output with intraluminal squalamine. A) PrP-A53T mice (black) had a significantly reduced total number of contractions compared to non-Tg (gray) at baseline and threshold. Threshold significantly increased the total number of contractions in both non-Tg and PrP-A53T mice (N = 6–12 mice/group, 1-way ANOVA). B) There was no difference in PCC velocity between PrP-A53T mice (black) compared to non-Tg (gray) at baseline and threshold. Intraluminal squalamine (30μM) significantly increased colonic PCC velocity in non-Tg (gray patterned) and A53T (black patterned) at baseline (N = 6–12 mice/group, 1-way ANOVA). C) Feeding of squalamine for 5 days increased fecal pellet output in non-Tg and A53T mice at several doses. (N = 10 mice/group/dose, 2-way ANOVA). All data represented as mean±S.E.M, *p < 0.05.
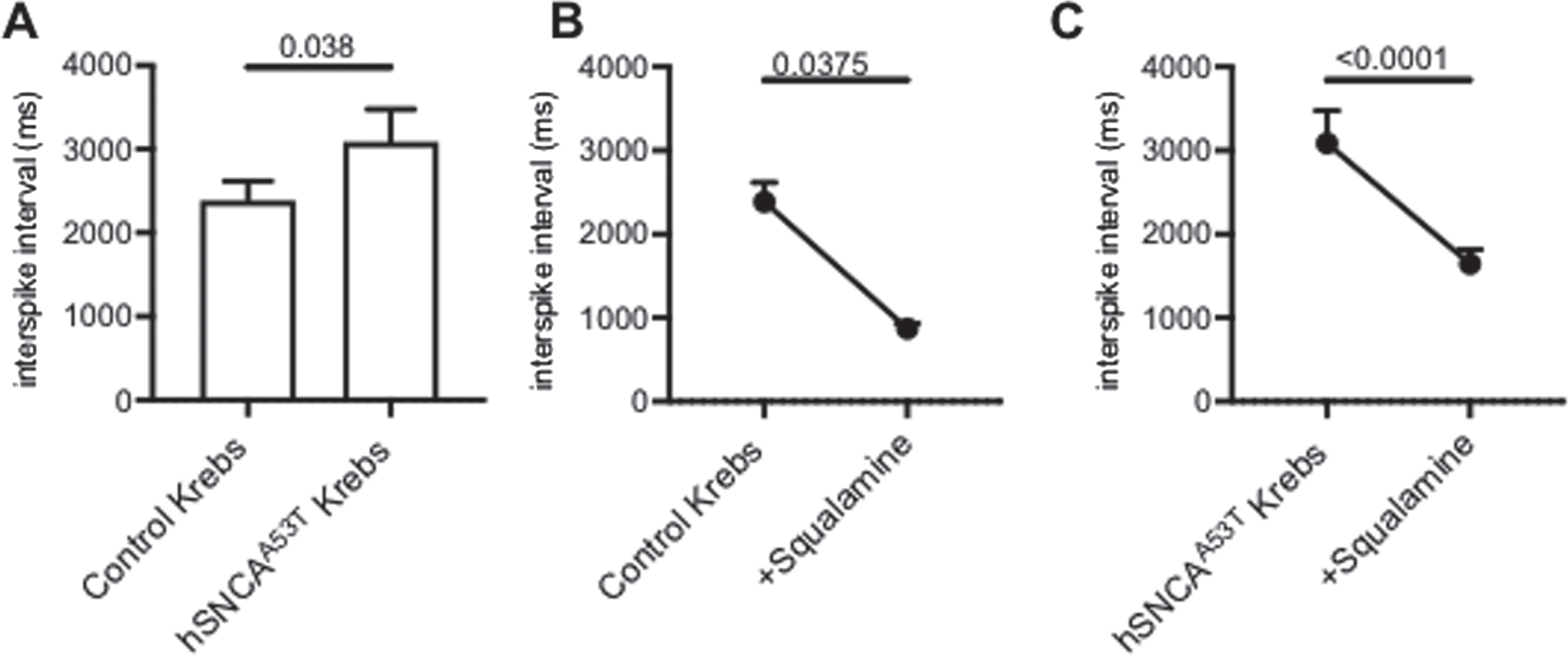
Intraluminal squalamine significantly increased vagal afferent firing (reduced interspike interval) in both control and hSNCAA53T mice. A) The interspike interval was significantly longer in hSNCAA53T mice than control mice (N = 13–14 mice/group). B) Intraluminal squalamine (30μM) significantly reduced the vagal afferent interspike interval for control mice (N = 13 mice). C) Intraluminal squalamine (30μM) significantly reduced the vagal afferent interspike interval for hSNCAA53T mice (N = 14 mice). 1-way ANOVA, Holm-Sidak’s multiple comparisons test. All data represented as mean±S.E.M, *p < 0.05.
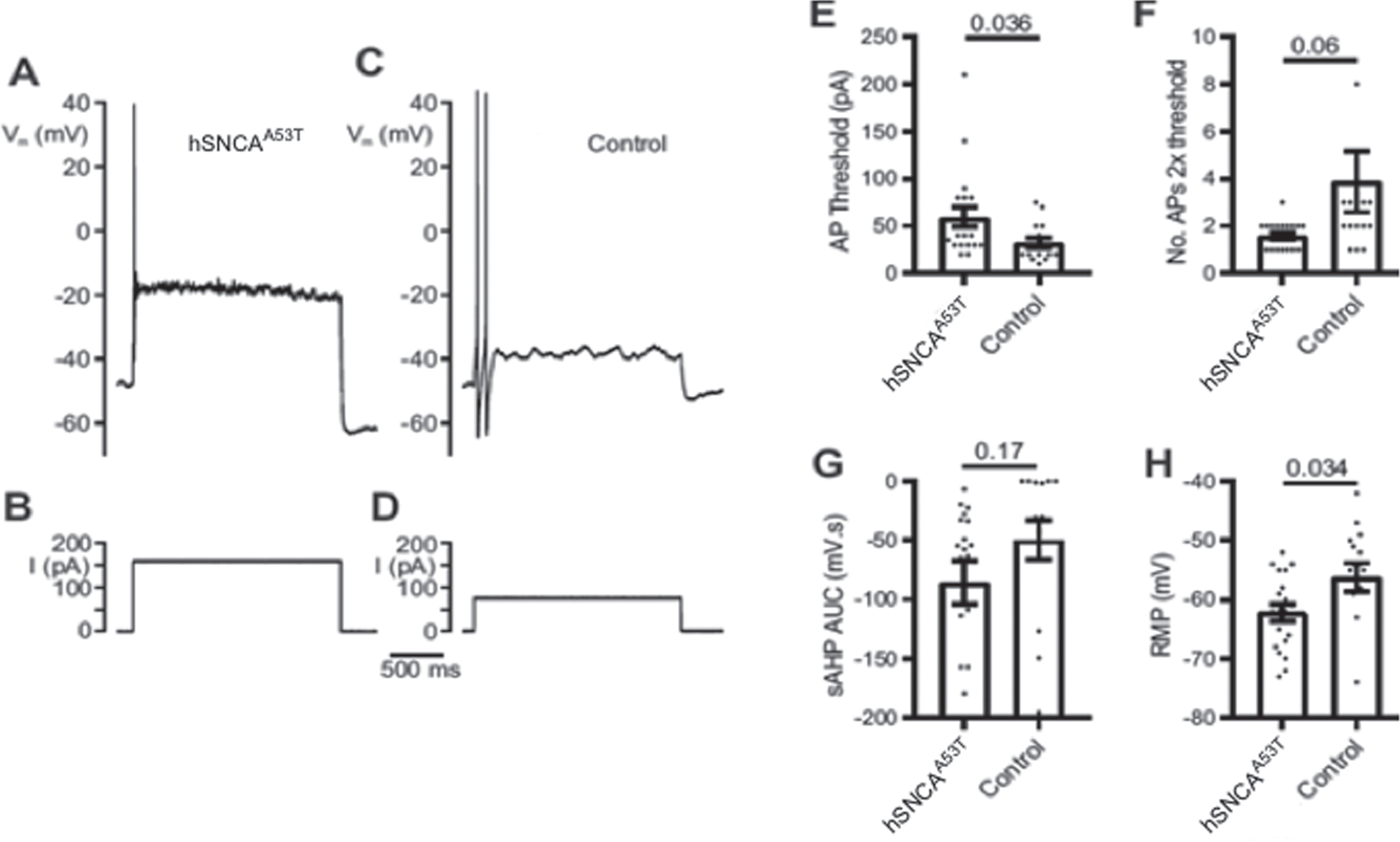
hSNCAA53T (endogenous promoter) mice had decreased intrinsic excitability of myenteric intrinsic primary afferent neurons compared to control mice. A) Representative action potential firing response to injected depolarizing square wave current stimulus (B) of 2× threshold intensity (hSNCAA53T). C) Representative action potential firing response to current stimulus (D) of 2× threshold intensity (control). E–H) Probabilities under the null hypothesis of no difference given above dot plots, mean values given by open bars, error bars represent SEM. E) The threshold intracellular current (AP threshold) required to evoke a single action potential (AP) was larger for hSNCAA53T (N = 20 recordings) than for control mice (N = 16 recordings) (t = 2.2, t-test unpaired 2-tailed). F) The number of action potentials evoked at 2 times threshold current intensity (No. APs 2× threshold) was greater for control (N = 19 recordings) than for hSNCAA53T mice (N = 16 recordings) (t = 1.9, t-test unpaired 2-tailed). G) The slow action potential after hyperpolarisation area under the curve (sAHP AUC) showed little difference between hSNCAA53T (N = 19) and control mice (N = 14) (t = 1.4, t-test unpaired 2-tailed). H) The resting membrane potential was more hyperpolarised for hSNCAA53T (N = 20) than for control mice (N = 16 recordings) (t = 2.2, t-test unpaired 2-tailed).
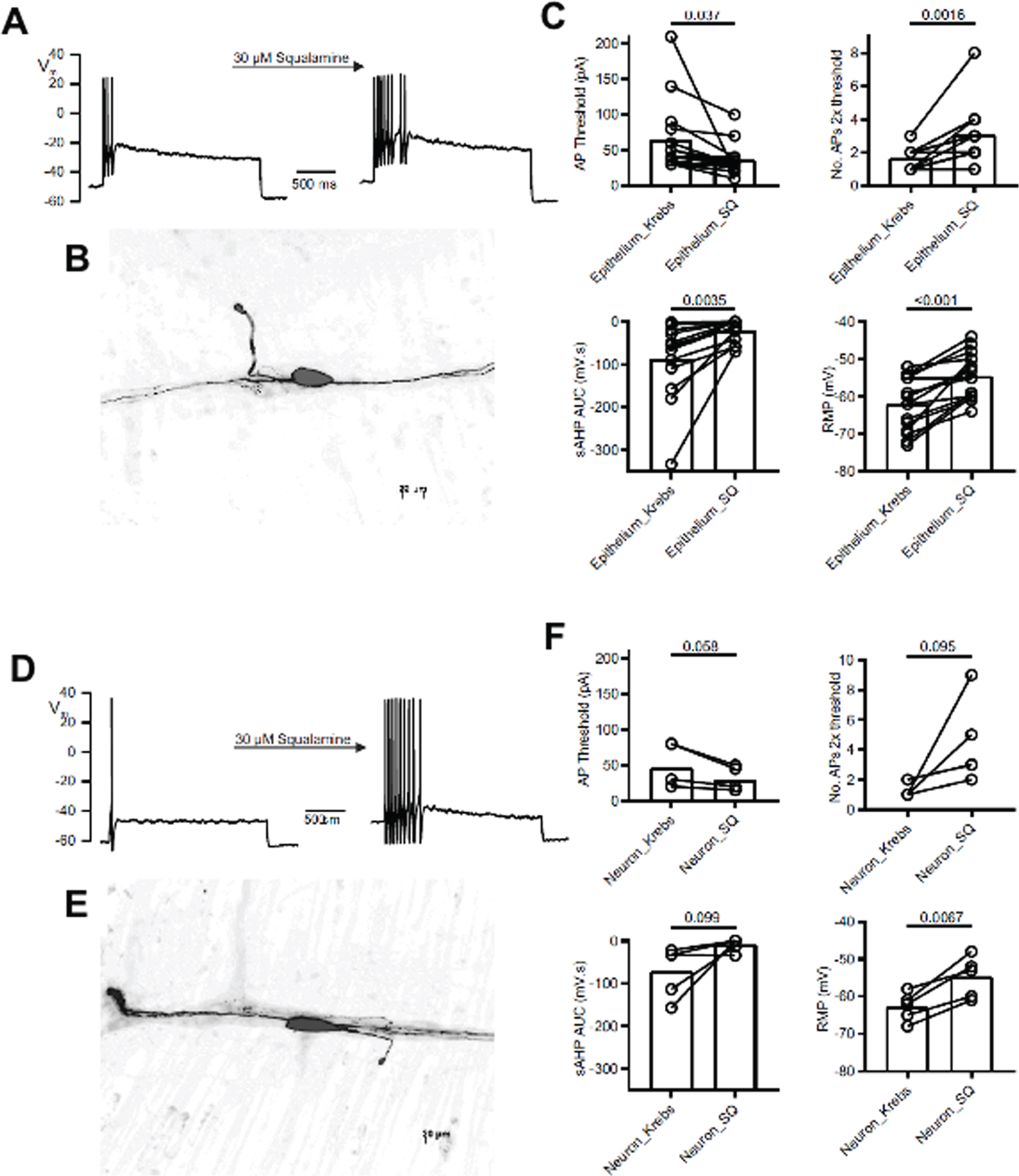
Application of squalamine onto the intestinal epithelium or directly onto the exposed myenteric plexus increased excitability of intrinsic primary afferent neurons (IPANs) in hSNCAA53T mice. A) Representative action potential firing increase to injected square wave current stimulus after acute application of 30μM squalamine onto the intestinal epithelium using the divided hemidissection preparation. B) Texas Red fluorescence image of neuron recorded from in (A) after tissue fixation revealing flattened oval soma and circumferentially directed neurites (Dogiel type II morphology) characteristic of chemosensitive myenteric intrinsic primary afferent neurons. C) Addition of squalamine to the epithelial layer decreased the action potential firing threshold (AP Threshold) (t = 2.3, t-test paired 2-tailed), increased the number of action potentials discharged (No. AP 2× threshold) (N = 15 recordings, t = 4, t-test paired 2-tailed), decreased the area under the curve for the post action potential slow after hyperpolarisation (sAHP AUC) (N = 14 recordings, t = 3.6, t-test paired 2-tailed) and depolarised the neuron membrane potential (N = 15 recordings, t = 5.9, t-test paired 2-tailed). D) A representative action potential firing increase in response to an injected square wave current stimulus after application of 30μM squalamine onto the myenteric plexus. E) Texas Red fluorescence image of neuron recorded from in (D) reveals Dogiel type II morphology. F) Addition of squalamine to the myenteric plexus decreased AP threshold (N = 5 recordings, t = 2.6, t-test paired 2-tailed), increased No. AP 2× threshold (N = 5 recordings, t = 2.2, t-test paired 2-tailed) decreased the post action potential sAHP AUC (N = 5 recordings, t = 2.1, t-test paired 2-tailed), and depolarised resting membrane potential (N = 5 recordings, t = 5.2, t-test paired 2-tailed). C, F) Probabilities under the null hypothesis of no difference given above individual value barbell plots, mean values given by open bars, error bars represent SEM.
7-month-old male and female PrP-A53T human α-syn overexpressing transgenic mice (prion promotor) and their non-Tg littermate controls (25–35 g) [29] were reared in a breeding colony at the Florey Institute. A total of 30 mice were used for in vitro colonic motility (Fig. 2A). For the dose ranging in vivo fecal pellet output test, a total of 100 male mice, or 5 sets of non-Tg (N = 10) and PrP-A53T (N = 10) mice aged 7-months were used.
All mice were housed 3–5 per cage on a 12 h light/dark cycle with food and water provided ad libitum and allowed a 1-week acclimation period after arrival.
Squalamine dilactate
Squalamine dilactate was donated by Dr Michael Zasloff, Georgetown University (Washington, DC, United States). Squalamine dilactate powder was dissolved in 90% ethanol to make a stock solution, then aliquoted and stored at –20°C until use. Stock solution was diluted in Krebs buffer to a working concentration of 30μM for in vitro experiments, as determined in a dose-ranging pilot experiment [30]. Squalamine was diluted to 20, 40, 80, or 120 mg/kg for oral gavage (2% ethanol, included in the control gavage), in order to study in vivo fecal pellet output.
In vitro colon motility
The in vitro colonic segment experiments were performed with only minor differences in equipment/protocol and as described previously [31–35]. The colon was excised and placed within an organ-bath perfusion chamber filled with warmed, oxygenated Krebs buffer or physiological saline (35°C, 95% O2, 5% CO2). The colon was flushed and cannulated at the oral and anal ends to a manifold and syringe to allow inflow of oxygenated Krebs buffer (or physiological saline) or Krebs and squalamine and to maintain intraluminal pressure. Recordings in the first portion of the study (data in Fig. 1) were measured at a mechanical threshold causing a pressure differential of 2 hPa. The height of the inflow tube at baseline measurements (data in Fig. 2A) was parallel to the height of the colon in the organ bath (1.1 cm). Mechanical threshold was defined as an inflow pressure great enough to generate a contraction in under 30 s (1.8 cm). Motor patterns were recorded using a Microsoft LifeCam 3000 web camera (Fig. 1) positioned 7–8 cm above the tissue. Videos were recorded during a 20 min Krebs control and a 20 min Krebs plus squalamine period in which solutions were added to the inflow syringe.
In vivo fecal pellet output (FPO)
PrP-A53T and non-Tg controls were subjected to the FPO test 1 day prior to the start of dosing (day 0). Mice were fasted for one hour and then given access to food one hour before testing. Baseline values were obtained on day 0, following oral gavage with vehicle (sterile water). On days 1–5 mice were fasted for 1 h prior to oral gavage with vehicle or 20, 40, 80, or 120 mg/kg squalamine. Oral gavage occurred between 10:00 to 11:00 am daily. On day 5, the FPO test was performed 1 h after the final dose was administered. Total number of stool pellets produced in the first 15 min and over a 60 min period was measured in each group.
Mesenteric nerve recording
Jejunal mesenteric nerve recordings were performed as described previously [21]. A segment of hSNCAA53T mouse jejunum (2–3 cm) was mounted on an agar-coated petri dish filled with oxygenated Krebs buffer and nicardipine (3μM) and the attached mesentery was pinned out. The luminal contents of the tissue were flushed with Krebs and the oral and anal ends of the tissue were cannulated with silicon tubing. The mesenteric nerve bundle was exposed using fine-tipped forceps by gently removing excess mesentery around the mesenteric arteries. The petri dish nerve preparation was then placed on an inverted microscope stage and perfused with fresh oxygenated Krebs in the serosal compartment using a pump. The nerve bundle was sucked onto a glass micropipette attached to a microelectrode to record multi-unit electrical activity recorded using a Multi-Clamp 700B amplifier and Digidata 1440A signal converter. Baseline afferent firing was recorded for 20–30 min during luminal Krebs perfusion. Intraluminal squalamine (30μM) was perfused following the control (Krebs) for a duration of 30 min. Single-unit activity was sorted from multi-unit recordings using principal component analysis (PCA) and spike waveform analysis for spike shape, size and duration in the Dataview program [36]. Vagal fibres were identified by response to cholecystokinin (CCK) applied to the serosa 10 min following the cessation of treatment [37, 38]. The mean interspike interval between vagal afferent spike firing (the inverse of firing frequency) was measured during Krebs and squalamine treatment periods for control and hSNCAA53T mice.
Whole-cell patch clamping
Whole-cell patch clamp was performed on a myenteric plexus preparation as previously described [39]. The mucosa, submucosa, and circular muscle were removed to expose the myenteric plexus over about half the length of the preparation while the adjacent epithelium remained intact. The preparation was dissected in a carbogen-bubbled recording dish filled with Krebs buffer. A single myenteric ganglion was cleaned for patching by application of 0.02% protease type XI (Sigma-Aldrich, Oakville, Canada) dissolved in Krebs for 10–15 min followed by washout with perfused Krebs for 5 min. Only whole-cell recordings greater than 4 GΩ were used. IPANs’ excitability parameters were measured as described previously first with perfusion with Krebs buffer for 20 min followed by perfusion with Krebs buffer containing squalamine for 15–30 min. The patched cells were confirmed to be IPANs at the end of the recording by injection of Neurobiotin and confirming the morphology of the cell was that of a Dogiel type II neuron [40]. The neurons were ionophoretically filled with Neurobiotin, from the intracellular patch pipette saline and then fixed overnight at 4°C in Zamboni’s fixative (2% vol/vol picric acid, 4% paraformaldehyde in 0.1 M Na2HPO4/NaH2PO4 buffer, pH = 7.0) [41]. Neurons were subsequently washed using three 10 min washes of DMSO and then PBS and then visualized with fluorescence epi-illumination after exposure to streptavidin-Texas Red (Vector, http://www.vectorlabs.com), diluted 1:50, revealing Neurobiotin [41, 42].
Statistical analysis
Effects of squalamine on IPANs’ excitability in control and PD model mice were assessed in paired experiments following Krebs control and subsequent squalamine exposure. Percent difference was calculated by (treatment-control)/control. Data are presented as mean±S.E.M. In vitro statistical comparisons were performed using paired or unpaired, two-tailed t-tests or 1 or 2-way ANOVA using Graphpad Prism software (Version 7.0). In vivo studies were analysed using 1-way and 2-way ANOVA. Statistical significance was determined when p < 0.05.
RESULTS
Squalamine increased colonic motor activity in hSNCAA53T mice, in vitro
Colonic segments from the homozygotic A53T human α-syn overexpressing mice (hSNCAA53T) (8–9 months) and their controls (8–9 months) were compared to assess the effect of α-syn aggregation on colonic motility. The velocity of propagating contractile clusters (PCCs) was not significantly different in colonic segments from control mice compared with hSNCAA53T mice (N = 12–13 mice/group) (p = 0.0946) (Fig. 1A). According to the 2-way ANOVA, there would be 9.5% chance of observing the present effect if the animal model had no effect at all, therefore the observed effect of the animal model was not quite significant. Addition of intraluminal squalamine (30μM) increased PCC velocity by 20% in the control (p = 0.044) and by 38% in hSNCAA53T mice (p < 0.0001), compared to Krebs. In contrast neither the frequency nor force (amplitude) of the PCCs differed significantly between control and hSNCAA53T intestinal preparations, nor did squalamine increase either parameter in control or hSNCAA53T intestinal segments (Fig. 1B, C). In addition, while the colonic segment from the control animals exhibited a regular period pattern of peristaltic waves, the colonic segments from the hSNCAA53T model exhibited an irregular peristaltic wave pattern characterized by discontinuous contractions, as shown in the spatiotemporal maps (Fig. 1D, E, top). Addition of intraluminal squalamine stimulated regular peristaltic waves that travelled the full length of the colonic segment from the oral to anal direction (Fig. 1D, E, bottom).
Colonic motor activity is reduced in a second mouse model (PrP-A53T), in vitro
We evaluated the total number of contractions in non-Tg and PrP-A53T mice at baseline and during pressure-induced distention (threshold) (Fig. 2A). We also evaluated the effect of intraluminal squalamine on PCCs when colon segments were not distended (baseline) and during pressure induced distension (Fig. 2B). The total number of contractions was significantly reduced between PrP-A53T and non-Tg mice both at baseline (1.3±0.4 mm/s compared to 4.7±0.9 mm/s, p < 0.01) and at threshold (5.5±1.6 mm/s compared to 13.4±1.4 mm/s, p < 0.01) (Fig. 2A) Threshold significantly increased the total number of contractions compared to baseline in both groups (p < 0.0001 in non-Tg and p < 0.05 in PrP-A53T). There was no difference in PCC velocity for segments from PrP-A53T (prion promotor) mice compared to non-Tg controls (N = 6–12 mice/group) at both baseline (1.2±0.2 mm/s compared to 1.7±0.3 mm/s) and upon distention (1.6±0.3 mm/s compared to 3.0±0.7 mm/s) (Fig. 2B). Intraluminal squalamine (30μM) increased baseline PCC velocity from 1.2±0.2 mm/s to 2.3±0.4 mm/s for PrP-A53T mice (p < 0.05) and from 1.7±0.3 mm/s to 2.8±0.4 mm/s in non-Tg mice (p < 0.05) (Fig. 2B). Upon colonic distension, squalamine had no effect on PCC velocity in PrP-A53T mice (from 1.6±0.3 to 2.1±0.3 mm/s) or non-Tg mice (from 3.0±0.7 to 2.4±0.3 mm/s) (p > 0.05). These experiments demonstrate that colonic motility was impaired in vitro in both hSNCAA53T and PrP-A53T models and that squalamine increased PCC velocity for both control and hSNCAA53T and PrP-A53T mice.
Oral administration of squalamine increased colonic transit in the PrP-A53T mice strain in vivo
Fecal pellet output following oral gavage with squalamine was measured in the PrP-A53T mice to assess repeated oral administration of squalamine in a dose ranging study on defecation, in vivo (Fig. 2C). Pellet output within the first 15 minutes following oral gavage provided an estimate of colonic motility, since gastric feeding stimulates colonic peristalsis, resulting in the discharge of fecal pellets. Both PrP-A53T and non-Tg mice (N = 10 mice/group/dose) were administered vehicle only on Day 0 to establish each animal’s baseline pellet output. From Day 1 to Day 5 the animals received vehicle only or squalamine orally by gavage at a range of daily doses from 0 (vehicle only), 20, 40, 80, and 120 mg/kg. Pellet output within the first 15 min at Day 5 were compared to baseline values measured at Day 0. We could not detect a significant difference in the fecal pellet output between non-Tg and PrP-A53T mice in the groups receiving only vehicle (p > 0.05), possibly due to the poor sensitivity of this assay when compared to measuring PCC velocity and frequency, in vitro. However, following gavage of squalamine on Day 5, a treatment associated increase was observed in non-Tg mice dosed with 40 and 120 mg/kg (p < 0.01 and p < 0.05, respectively) as well as in PrP-A53T mice dosed with 20, 40, and 80 mg/kg (p < 0.005, p < 0.0001, and p < 0.01, respectively). These experiments demonstrated that oral administration of squalamine stimulated colonic motility in both non-Tg and PrP-A53T mice. Effect of squalamine on wet weight, dry weight, % water content of stool, and % change in stool output/body weight are presented in Supplementary Figure 1.
Squalamine significantly increased vagal afferent firing frequency in hSNCAA53T mice in vitro
Vagal afferent firing rates were measured by extracellular action potential (multiunit) recording from a mesenteric nerve bundle attached to segments of the jejunum excised from hSNCAA53T mice and controls. The interspike interval of vagal single units were measured and these represent the reciprocal of their firing rate. Mean afferent vagal firing rate was significantly reduced in the hSNCAA53T mice compared to the control group (p = 0.0375). The mean interspike interval between vagal spikes was shorter in control mice compared to hSNCAA53T mice, indicating a lower firing rate in the latter (2389±230 ms compared to 3088±392 ms) (Fig. 3A). Squalamine had strong effects on vagal firing rates in both groups of mice (Fig. 3B, C). Intraluminal addition of squalamine (30μM) decreased the interspike interval between vagal afferent spikes in the control mice by 63% to 874±60 ms (p < 0.0001). Acute treatment with squalamine also decreased the interspike interval between vagal afferent spikes in the hSNCAA53T model mouse by 47% to 1646±168 ms (p < 0.0001). These results demonstrate that the vagal afferent firing rate is reduced in the hSNCAA53T mice and is potently stimulated by the addition of intraluminal squalamine in both the control and hSNCAA53T model mice.
hSNCAA53T model mice have reduced myenteric intrinsic primary afferent neuron (IPANs) excitability
To help elucidate the mechanism by which squalamine stimulated intestinal motility we conducted electrophysiological studies on single neurons within the intact myenteric plexus of the hSNCAA53T mice [27] and corresponding control animals using published methods [18]. IPANs [40] in the myenteric plexus have neurites that project to the epithelial layer where they respond to molecules present in the gut lumen and then conduct action potentials to the myenteric plexus [39, 44]. IPANs excitation generates PCCs that move luminal contents in the oral to anal direction [44]. We investigated whether IPANs’ excitability was reduced in the hSNCAA53T mice compared to the control mice and whether squalamine administration to intestinal segments taken from hSNCAA53T mice could facilitate IPANs’ excitability.
Using whole-cell patch pipette recordings from IPANs, we measured the threshold for action potential generation in response to intracellular injection of square depolarising current pulses (AP threshold), the number of action potentials (AP) generated in response to current injection of 2× threshold intensity (No. AP 2× threshold), the area under the curve for the slow after-hyperpolarisation generated by 3 action potentials (sAHP AUC), and the resting membrane potential. In this experiment, 14 IPANs from 14 hSNCAA53T mice and 9 IPANs from 9 control mice were successfully studied (Fig. 4). The AP threshold (mean±SEM) was 32.2±20.0 pA for the control compared to 59.2±46.1 pA for the hSNCAA53T IPANs (p = 0.036) (Fig. 4E). The number of action potentials produced by a current 2× the threshold intensity was 3.9±5.1 for the control versus 1.6±0.6 for the hSNCAA53T IPANs (p = 0.06) (Fig. 4F). The area under of the curve for the sAHP was –49.5±63.7 mV s for the control versus –85.5±78.2 mV s for the hSNCAA53T cells (p = 0.17) (Fig. 4G). The resting membrane potential was –56±10 mV for control IPANs versus –62±6 mV for those from the hSNCAA53T model (p = 0.034) (Fig. 4H). Thus, IPANs from the hSNCAA53T mouse strain exhibited a reduced excitability compared to those from the control animals.
Squalamine excites IPANs
We tested for the effect of squalamine on the excitability of an isolated colonic segment from the hSNCAA53T (endogenous promoter) mouse using preparations in which myenteric neurons are exposed on one half of a small intestinal segment [39]. In this experiment we asked whether squalamine influenced the activity of the IPANs through direct interaction with the neuron or indirectly, by stimulating release of epithelial mediators that influenced IPANs’ behaviour. Addition of squalamine (30μM) to the Krebs buffer in either the epithelial or the myenteric plexus compartments of the preparation increased IPANs’ excitability (Fig. 5). Adding squalamine to the luminal side of the hSNCAA53T mouse colon (N = 15 cells) decreased the mean AP threshold from 63.7±50.4 to 35.7±22.3 pA (p = 0.037) (Fig. 5C) and increased the number of APs produced by a current 2× the threshold intensity from 1.6±0.6 to 3.1±0.7 (p = 0.0016) (Fig. 5A,C). Addition of squalamine decreased the area under the curve of the sAHP from 86.8±88.2 to 20.3±25.3 mV s (p = 0.0035) (Fig. 5C) and depolarised the resting membrane potential from –62±7 to –54±6 mV (p < 0.001) (Fig. 5C). Adding squalamine directly to the myenteric plexus of the hSNCAA53T mouse colon (N = 5 cells) (Fig. 5D,E) did not significantly decrease mean AP threshold from 46.0±31.3 to 29.0±10.1 pA (p = 0.058) and increase the number of APs produced by a current 2× the threshold intensity from 1.4±0.5 to 4.4±2.8 (p = 0.095). Squalamine significantly depolarised the resting membrane potential from –63±4 to –55±6 mV (p = 0.0067), but did not significantly decrease the area under the curve of the sAHP (–71.9±60.1 to –9.6±15.1 mV s (p = 0.099)), when added to the myenteric plexus of the hSNCAA53T mouse. These experiments demonstrated that squalamine can augment the reduced excitability of the IPANs in tissue taken from hSNCAA53T mice and that squalamine can act directly on the IPANs, rather than indirectly through release of a chemical such as a neurotransmitter from a luminal epithelial cell, like an enteroendocrine cell.
DISCUSSION
The purpose of the present study was to investigate the ability of squalamine, a zwitterionic amphipathic aminosterol that has previously been shown to improve age-related gut dysmotility [25, 30] and inhibit the formation of α-syn aggregates [24], to improve colonic motility and constipation in two mouse models of PD. We demonstrate in this report that squalamine can restore gastrointestinal motility in mouse models of PD.
Constipation in PD presents a significant challenge in the management of the disease and often precedes the onset of motor symptoms by years or decades [9]. It has been recently reported that α-syn expression is induced in the ENS in response to viral, bacterial and fungal infections [45] and that excessive intraneuronal accumulation of α-syn promotes formation of toxic aggregates [24]. Because of the normal trafficking of α-syn aggregates from the ENS to the central nervous system (CNS) via the vagus [10, 16], neurotoxic aggregates accumulate progressively within the brainstem and more rostral structures. Additionally, there is potential for bidirectional spread of α-syn between the ENS and CNS via efferent vagal fibres, as suggested by Di Monte [46]. α-syn overexpression in the DMV and vagal ganglia was efficiently trafficked via vagal afferents and efferents to the stomach wall [46]. It would be logical to suggest therefore that targeting α-syn aggregation in the ENS may be beneficial in the treatment of PD constipation [47].
The two models of PD used in this particular study both expressed the human A53T α-syn autosomal dominant mutation, one being driven by a prion promoter (PrP-A53T) [29], the other by the endogenous mouse sequence (hSNCAA53T) [27] and both exhibit GI dysmotility. Overexpression of normal human α-syn in the ENS of the control strain was not associated with apparent gastrointestinal dysfunction, nor were α-syn aggregates seen on pathology, in contrast to the strain engineered to overexpress A53T (hSNCAA53T) [27]. Addition of squalamine increased colonic PCC velocity in both strains and FPO in the PrP-A53T strain. It is notable however, that squalamine also produced prokinetic effects on motility in control strains in the absence of α-syn pathology, which likely occurs via stimulation of the ENS. These observations support the hypothesis that the beneficial effect of squalamine in PD model mice with, respect to motility, may in part involve displacement of membrane disruptive aggregates within the enteric neurons.
Indeed, squalamine has been shown to have two beneficial properties of relevance to PD. By displacement of monomeric α-syn from a membrane, squalamine reduces the surface concentration of the protein, reducing the probability that neurotoxic aggregates will form. In addition, squalamine can also displace aggregates that have already formed on the membrane, restoring membrane function, and releasing the aggregate into the cytoplasm for subsequent digestion. Upon entry into a cell, or potentially a primary afferent neuron [48], squalamine is electrostatically attracted to the negatively charged phospholipid head groups that comprise the cytoplasmic surface of the plasma membrane thereby neutralizing its negative electrostatic surface potential [48, 49]. In a C. elegans model of PD, in which the animal has been engineered to express human α-syn in its muscles, orally administered squalamine inhibits formation of membrane disruptive intracellular aggregates and prevents the onset of disordered motility and paralysis [24]. In addition, in vitro, squalamine prevents the binding of membrane disruptive α-syn aggregates to the surface of neuronal cells, protecting them from loss of viability [24]. We believe a similar effect occurs when squalamine enters the enteric neuron of the hSNCAA53T mouse. Our studies demonstrate, as well, that the enteric nerves in the hSNCAA53T mouse models are not irreversibly damaged, since squalamine restored the disordered GI motility in both models of PD. Orally administered squalamine also increased FPO in A53T mice at the mid-range doses of 20, 40, and 80 mg/kg and at the doses of 40 and 120 mg/kg in the non-Tg controls, which may represent a bell-shaped dose response curve for squalamine, with an optimal range of 40 mg/kg. Additionally, chronic administration may produce significant effects on α-syn aggregates by day 5. Indeed, in a recent Phase 2a clinical study of 50 PD patients with severe constipation, orally administered ENT-01 reversed constipation dose dependently in most patients some suffering for 60 years [26].
As predicted in the introduction, constitutive vagal afferent firing rates were decreased (increased interspike intervals) for hSNCAA53T model animals compared to controls (Fig. 3). Squalamine (30μM) potently increased vagal fibre firing rates when it was added to the Krebs buffer perfusing the lumen for both controls and hSNCAA53T animals. Given the important role of enteric IPANs in gut to brain signaling [21], the vulnerability of the ENS to misfolded α-syn in terms of reduced function or IPANs excitability would have a significant impact on the amount and quality of information reaching the brain from the gut. If behavioural or mood-related Parkinsonian symptoms are mediated, in part, by reduced afferent vagal discharge, the excitatory action of squalamine on IPANs and the vagus may serve to reverse the symptoms.
We have shown here that IPANs from animals in which A53T is overexpressed (hSNCAA53T) exhibit reduced excitatory activity compared with IPANs from controls. Reduced excitability was characterized by a more hyperpolarized resting membrane potential in the hSNCAA53T mouse, requiring a larger threshold current for action potential generation. The number of action potentials produced by a current 2× the threshold intensity was much lower for the hSNCAA53T mouse, which correlates with the larger area under the curve of the slow after-hyperpolarization (sAHP). These results demonstrate that the IPANs from the hSNCAA53T mouse requires a greater stimulus to fire an action potential and takes longer to repolarize before it can fire a subsequent action potential (AP) than the control mouse and is thus less excitable.
Subsequent squalamine exposure restores normal excitatory responses of the colonic IPANs within minutes of local application to preparations from hSNCAA53T animals; the resting membrane potential is more depolarized. In this model less current is required to reach threshold and generate an AP, and the sAHP area under the curve (AUC) is smaller, indicating faster repolarization and the ability to respond and produce a subsequent AP sooner. It is possible that the effects on PD model mice are not α-syn specific, given the prokinetic effects of squalamine in young and old wild-type mice [25] and in the controls. In the setting of the GI tract, we hypothesize that squalamine can be readily transported across the intestinal epithelium and, as described in this report, stimulate the activity of local IPANs and, in turn, GI motility in a healthy animal. It is also possible that IPANs are stimulated via a sensory transduction cascade mediated by intestinal epithelial cells but given that squalamine increases IPAN excitability when applied luminally or directly to the myenteric plexus, we suggest the former. Interestingly, squalamine does not seem to enter the systemic circulation so that the effects of oral administration are focused in the gut [26].
Locally applied squalamine can stimulate the peristaltic activity of both WT and A53T mice minutes after exposure of the colonic lumen to the compound. Similarly, squalamine restores normal excitatory responses of the colonic IPAN within minutes of local application to preparations from both control and A53T animals. These observations are consistent with a mechanism in which the squalamine ion increases the excitatory status of the IPAN, regardless of the presence of functionally disruptive α-syn aggregates. A reduction in the negative surface charge on intracellular membranes could both influence the electrical behavior of the neuron as well as displace membrane associated aggregates of α-syn. Because squalamine can displace proteins that are bound electrostatically to intracellular membranes, it rapidly activates the AMPA glutamate receptor in primary cortical neurons by displacing the inhibitory TARP regulatory protein from the cytoplasmic face of the plasma membrane [48]; similarly the squalamine ion inhibits the sodium hydrogen exchanger (type 3) by displacing its positively charged carboxyl-terminus from the cytoplasmic face of the plasma membrane [50]. The mechanism by which squalamine alters the electrical excitability of the enteric neuron, independent of the presence of α-syn, is under investigation.
The action of squalamine on IPANs to increase their excitability is entirely consistent with its prokinetic effects since propulsive motility throughout the small and large intestines is critically dependent on normal IPANs function and excitability [20, 51]. Silencing of IPANs by inhibition of protein kinase A activity produces lethal pseudo-obstruction in the murine intestine [20], and the pharmacological inhibition of IPANs excitability by application of 5,6-Dichloro-1-ethyl-1,3- dihydro-2H-benzimidazol-2-one to the Krebs buffer superfusing ex vivo segments of mouse intestine reduces and then abolishes all propulsive PCCs [51]. Manipulation of the current underlying the AHP by calcium-activated, intermediate-conductance potassium channels (IK or IKCa) block or activation has similar effects in the small intestine [52]. Because of their role in generating the propulsive peristaltic reflex, action at myenteric IPANs provides a cellular explanation for the increased propulsive motility and reduced constipation in the hSNCAA53T and PrP-A53T animals caused by squalamine. We found evidence for the direct action of squalamine on the IPANs as demonstrated by the increased excitability of the neuron when squalamine was applied directly to the myenteric plexus.
In conclusion, we have shown that squalamine, locally administered to the lumen of the GI tract of hSNCAA53T mice, can restore intestinal motility and enteric sensory neuron function, suggesting that the ENS is not irreversibly damaged, at least in murine models of PD.
CONFLICT OF INTEREST
M.Z. and D.B. are employees of Enterin Inc. J.B. and W.A.K. are members of the scientific advisory board of Enterin Inc. The authors have no additional conflicts of interest.
Footnotes
ACKNOWLEDGMENTS
This study was supported by grants from Enterin Inc. and the Natural Sciences and Engineering Research Council of Canada Discovery Grant (2014-05517) awarded to W.A.K. Takeda Pharmaceuticals, Inc. provided support to the experiments conducted in the Florey Institute.