
Abstract
The gut microbiome has been increasingly implicated in Parkinson’s disease (PD); however, most existing studies employ bacterial-specific sequencing, and have not investigated non-bacterial microbiome constituents. Here, we use fungal-specific internal transcribed spacer (ITS)-2 amplicon sequencing in a cross-sectional PD cohort to investigate associations between the fungal gut microbiome and PD. Fungal load among participants was extremely low, and genera identified were almost exclusively of proposed dietary or environmental origin. We observed significantly lower fungal DNA relative to bacterial DNA among PD patients. No fungi differed in abundance between patients and controls, nor were any associated with motor, cognitive, or gastrointestinal features among patients.
INTRODUCTION
Strong links between the gastrointestinal (GI) tract and Parkinson’s disease (PD) have long been recognized [1]. The high prevalence and early incidence of symptoms including constipation, delayed intestinal transit time, and small intestinal bacterial overgrowth; the detection of α-synuclein aggregates in the enteric nervous system; and the reduced lifetime risk of PD following full truncal vagotomy all support the hypothesis that PD pathophysiology may begin in the gut [1, 2]. Therefore, recent attention has turned to the potential role of the gut microbiome in this disease. Since 2014, numerous studies have reported gut microbiome alterations in PD [3, 4]. However, the vast majority have employed 16S rDNA amplicon sequencing which only captures the bacterial component of the microbiome. To date, a potential role for the fungal constituents of the gut microbiome, also known as the “mycobiome”, has remained unexplored.
Intriguing links exist between fungi and PD. Seborrheic dermatitis, a skin condition caused by overgrowth of the fungus Malassezia, is strongly linked with PD [5]. Levodopa has been shown to stimulate hyphal growth in Malassezia, suggesting that if this organism entered the CNS it would be most invasive in dopamine-rich brain regions, including the substantia nigra [5]. Furthermore, fungal DNA [6] and proteins [6, 7] have been detected in postmortem PD brains. Gut mycobiome dysbiosis has been observed in GI diseases such inflammatory bowel disease (IBD) and colorectal cancer [8–10]; a higher ratio of Basidiomycota:Ascomycota has been observed in both conditions [9, 10], and increased abundance of Candida albicans and higher overall fungal load have been observed in IBD [9]. However, whether there is a stable population of colonized fungi in the human GI tract in the absence of disease remains controversial [8, 11]. Given the GI dysbiosis observed in PD [1, 13], and the potential associations between skin- and CNS-relevant fungi in this disease, we hypothesized that gut mycobiome dysbiosis may be an unrecognized component of PD.
METHODS
Participant recruitment
Ninety-five PD patients and 57 controls were enrolled from the Pacific Parkinson’s Research Centre (PPRC) at the University of British Columbia (UBC), Canada. Bacterial microbiome results in this cohort have recently been published [14]. Inclusion/ exclusion criteria are described in the Supplementary Material. All participants provided written informed consent. Participant demographics are shown in Table 1.
Study participant demographics and characteristics. Values are presented as median (interquartile range), or percent. Differences in participant groups were assessed using Mann-Whitney U tests for numeric variables, and Fisher’s exact tests for categorical variables
Data and sample collection
Participants completed a 2-hour study visit at the PPRC. PD symptoms were assessed in the ON state using the Movement Disorder Society Unified Parkinson’s Disease Rating Scale (MDS-UPDRS). Cognition, depression, anxiety and fatigue were assessed via the Montreal Cognitive Assessment, Becks Depression Inventory-II, State-Trait Anxiety Inventory, and Fatigue Severity Scale, respectively. Irritable bowel syndrome and constipation were diagnosed by Rome III Functional Gastrointestinal Disorders criteria. Stool consistency was classified using the Bristol Stool Scale. A single fecal sample was collected into OMNIgene-GUT Kit tubes (DNA Genotek OMR-200) by participants at home. The tubes in these kits contain a stabilization buffer which halts microbial growth and stabilizes DNA, providing a snapshot of the microbial community at the time of collection. Samples were returned by mail, aliquoted, and stored at –80°C. Microbial DNA was extracted using QIAamp PowerFecal DNA Kits (QIAGEN 12830).
Fecal fungal sequencing
The fungal-specific internal transcribed spacer (ITS)-2 variable region was amplified with indexed ITS86F/ITS4R primers [15] on an Illumina MiSeq platform with 300 bp paired-end reads. Please see the Supplementary Material for additional sequencing details. Reads were processed using DADA2 in QIIME2 (v2019.7) [17]. Taxonomic classification was performed using the UNITE v04.02.2020 dynamic developer sequences [18]. Samples with < 100 reads post-filtering were not included in further compositional analysis. Alpha-diversity (Shannon) and beta-diversity (Bray-Curtis) were assessed using phyloseq (v1.26.0) [19] in R (v3.5.1) at a rarefaction depth of 978. Differential abundance analysis was performed with DESeq2 (v1.22.1) [20] and figures were created using ggplot2 (v3.2.1). Raw sequencing reads have been deposited to NCBI’s SRA, accession PRJNA664779.
Fungal load quantification
Total fungal load in participant fecal samples was quantified via qPCR using the same fungal-specific ITS2 primers used for sequencing, without barcodes and adapters. 10 ng of DNA was used per reaction. Total ITS2 counts were extrapolated from a standard curve of 107–101 ITS2 copies per reaction prepared from Saccharomyces cerevisiae genomic DNA. For normalization, total 16S was quantified using universal 16S 340F/514R primers (F: ACTCCTACGGGAGGCAGCAGT, R: ATTACCGCGGCTGCTGGC) against a standard curve of 107–101 16S copies per reaction from Escherichia coli genomic DNA. Although many samples had undetectable levels of fungal ITS2 DNA, bacterial 16S DNA amplification was highly robust for 100% of samples (CT range 11.4–13.7), demonstrating successful extraction of microbial DNA and an absence of PCR inhibitors in the eluted samples.
Statistical analysis
Differences in clinical characteristics between groups were assessed by Mann-Whitney U tests for numeric variables and Fisher’s exact tests for categorical variables. All correlation analyses employed Spearman correlation. In all cases of multiple hypothesis testing, Benjamini-Hochberg false discovery rate (FDR) correction was used, with a significance threshold of FDR-corrected-p < 0.05.
RESULTS
Fungi were extremely sparse among participants’ fecal microbiomes. After filtering, 106/152 participants (64/95 PD and 42/57 control) remained for downstream compositional analysis; the remainder had virtually no detectable fungal genomic content (<100 classifiable reads). In total, 503 unique amplicon sequence variants (ASVs) were detected, representing a total of 169 named genera. At the phylum level, 86% of reads mapped to Ascomycota and 12% to Basidiomycota (PD patients: 87% and 11%; controls 85% and 13%, respectively), with no difference in these phyla (nor the ratio of Ascomycota:Basidiomycota) observed between patients and controls. The most abundant fungal genera are presented in Fig. 1A; and distribution of the 6 most abundant genera in Fig. 1B. Saccharomyces was the most common genus among samples included in compositional analyses, at a mean relative abundance of 58.7%, and present in 94% of participants. Candida was present in 35% of participants, and Cladosporium and Penicillium each in 23%. No other genus was present in >20% of participants.
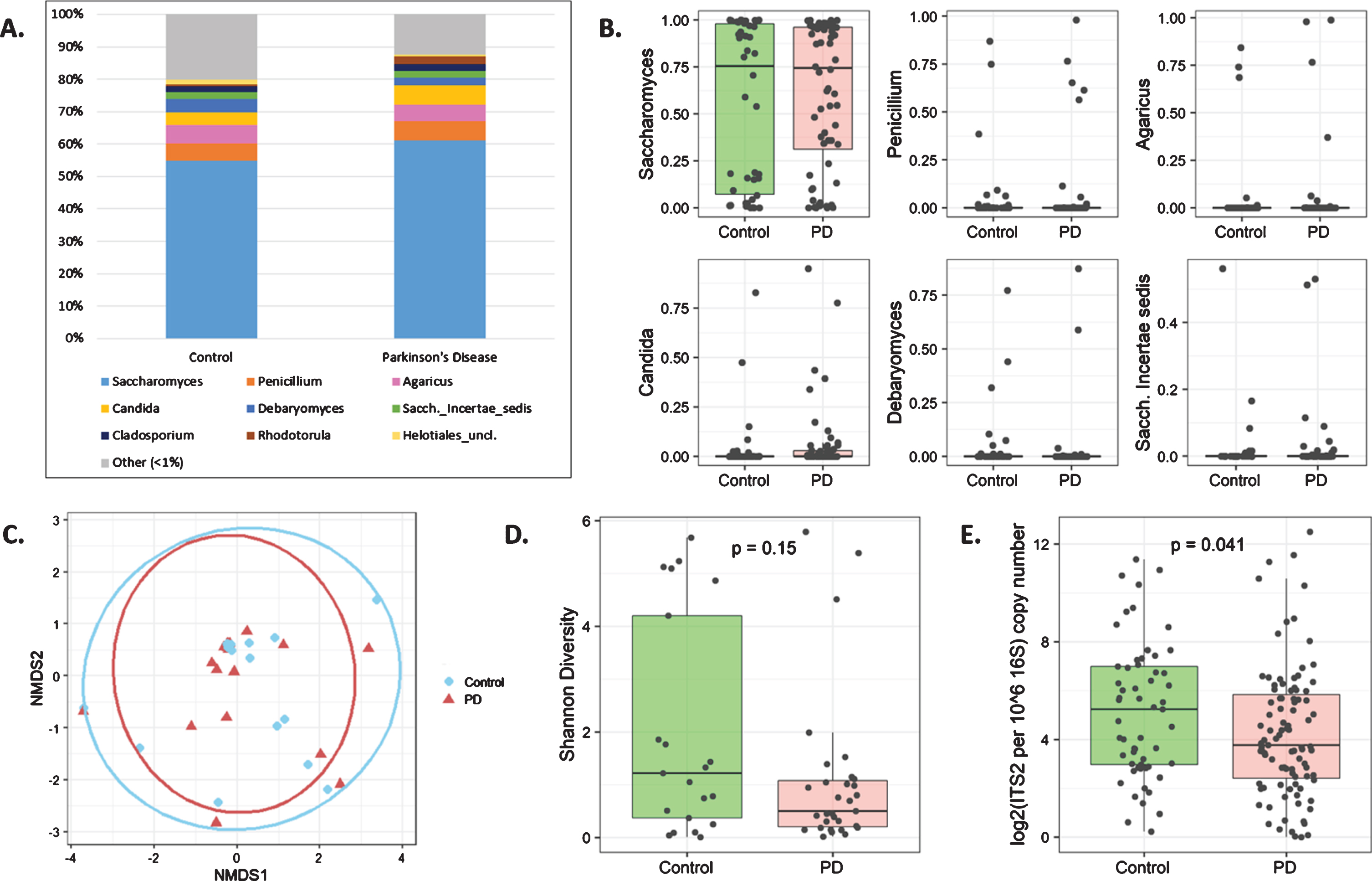
A) Stacked barplot of the top 9 most abundant fungal genera in controls and PD patients. B) No fungal taxa differed between PD patients and controls; relative abundances of the 6 most abundant genera are shown (all p-adj>0.99, DESeq2 analysis). C) No differences between participant groups were observed on a non-metric multi-dimensional scaling (NMDS) plot based on Bray-Curtis dissimilarity index. D) A trend towards lower fungal diversity (Shannon diversity index) was observed in PD. Only 31 PD patients and 21 controls were included in diversity analyses (D and E) based on a rarefaction depth of 987. E) Total fungal (ITS2) load relative to bacterial (16S) load was significantly reduced in PD patients.
No taxonomic differences were observed between patients and controls (DESeq2), nor was there a difference based on Bray-Curtis dissimilarity index (Fig. 1C). However, there was a non-significant trend towards lower fungal diversity in PD (Fig. 1D) and a reduced load of fungal ITS2 DNA relative to bacterial 16S DNA among patients (Fig. 1E).
We examined potential correlations between the abundance of specific fungal genera as well as total fungal load among PD patients and found no significant correlations with: non-motor symptoms, motor symptoms, motor scores, and motor complications (MDS-UPDRS parts I–IV respectively), total MDS-UPDRS score, Levodopa dose, cognition, depression, anxiety, or fatigue scores (Spearman correlations). Likewise, fungal composition and total fungal load did not differ by sex, constipation status, or use of any PD-modifying medications or laxatives (Mann-Whitney U tests) nor by Bristol score (ordinal logistic regression).
We recently published on bacterial microbiota dysbiosis in this same cohort [14]; using this data, we assessed whether there was any link between gut fungi and the bacterial microbiome. No significant correlations between any bacterial and fungal genera in participants were detected (all FDR-corrected p > 0.58). No bacteria were correlated with total fungal load.
DISCUSSION
Here, we report no significant differences in the gut mycobiome between PD patients and non-PD controls, and find no evidence of gut-fungal association with relevant disease parameters. The only significant observation between groups was a lower ratio of fungal to bacterial DNA among PD patients. This may be a denominator effect, as small intestinal bacterial overgrowth is common in PD [1]. However, it may also indicate an intestinal environment in PD that is less hospitable to eukaryotic cells.
Overall, the abundance and diversity of fungi in this cohort was very low. Other human mycobiome studies in healthy adults have reported similar findings, including that the mycobiome is dominated by yeast, the near-universality of Saccharomyces, low fungal diversity, and high inter-individual variability [8, 21]. In our study 98% of sequences mapped to two Phyla, Ascomycota (86%), and Basidiomycota (12%), which is nearly identical to results seen in a large healthy Human Microbiome Project cohort [8]. Malassezia, a predominantly skin-related genus that has been associated with PD, was very sparse in this cohort. Several species of skin-dwelling Malassezia have been identified [22], with M. globosa being reported as most prevalent in PD [22]; our amplicon-based sequencing did not provide species-level resolution. However, the overall low abundance and near-identical prevalence of Malassezia in patients and controls suggests distinct roles for this genus on the skin and in the gut in the context of PD. As ITS2 sequencing only assesses DNA, whether the fungi detected represent true colonized communities or simply transient cells derived from diet or environmental exposure is difficult to ascertain. Temporal results indicate that the mycobiome is highly unstable [8, 21], and it has been shown that most fungal DNA in the GI tract is also found in the oral cavity [11], suggesting that most intestinal fungi are derived from dietary and oral sources and likely do not exhibit robust colonization in the absence of disease. Indeed, all of the most-abundant genera detected in our cohort are common food-related or environmental fungi, including Saccharomyces, Penicillium [23], Agaricus [24], Candida [25], and Debaryomyces [23]. Furthermore, of these genera, only certain members of Saccharomyces and Candida are able to grow at body temperature [23]. Therefore, we suggest that the fungal constituents of the PD gut are more representative of the transient, unstable, non-colonizing fungi found in “healthy” guts rather than the fungal dysbiosis observed in diseases such as IBD and colorectal cancer [8].
While this is the first study to examine the mycobiome in PD, it has limitations that will need to be addressed in future work. Only one ITS2 primer pair was used for sequencing; primer efficiency and bias may be improved using other primers targeting different ITS regions [15]. Likewise, a multi-species mock community control would have allowed us to assess extraction and sequencing bias in our study. Data on relevant environmental factors (e.g., presence of mold in participant homes) were not collected. Although diet was assessed, it was in the form of an annualized food frequency questionnaire and we did not detect any correlations between dietary items and fungal genera; collecting data on food consumed specifically over the previous 1–2 days may be more informative in this regard.
In conclusion, we find no compelling evidence of gut mycobiome contribution to PD pathophysiology. This does not rule out a potential role for transient fungal dysbiosis contributing to PD etiology at the time of PD onset; large prospective studies would be needed to investigate this. Other often-ignored components of the gut microbiome, especially the virome, also deserve further investigation for a potential role in the etiology and pathology of PD.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
Footnotes
ACKNOWLEDGMENTS
This work was supported by Parkinson Canada; the Canadian Institutes of Health Research; and the Pacific Parkinson’s Research Institute. We thank Dr. Seti Boroomand and Faezeh Kharazyan from the Borgland Family Brain Tissue and DNA Biobank, at the Djavad Mowafaghian Centre for Brain Health, for their invaluable assistance. We are grateful to all individuals who participated in this study.