
Abstract
Background:
Recent evidence suggests that glucosidase beta acid (GBA) mutations predispose Parkinson’s disease (PD) patients to a greater burden of cognitive impairment and non-motor symptoms. This emerging knowledge has not yet been considered in patients who have undergone deep brain stimulation (DBS); a surgery that is generally contraindicated in those with cognitive deficits.
Objective:
To explore the long-term phenotypic progression of GBA-associated PD, in a DBS cohort.
Methods:
Thirty-four PD patients who had undergone DBS surgery between 2002 and 2011 were included in this study; 17 patients with GBA mutations were matched to 17 non-carriers. Clinical evaluation involved the administration of four assessments: The Mattis Dementia Rating Scale was used to assess cognitive function; non-motor symptoms were assessed using the Non-Motor Symptom Assessment Scale for PD; quality of life was measured using the Parkinson’s Disease Questionnaire; and motor symptoms were evaluated using part III of the Movement Disorders Society Unified Parkinson’s Disease Rating Scale, in on-medication/on-stimulation conditions. Levodopa equivalent doses (LED) and DBS settings were compared with clinical outcomes.
Results:
At a mean follow-up of 7.5 years after DBS, cognitive impairment was more prevalent (70% vs 19%) and more severe in GBA mutation carriers compared to non-carriers (60% vs 6% were severely impaired). Non-motor symptoms were also more severe and quality of life more impaired in GBA-associated PD. Motor symptoms, LED, and stimulation settings were not significantly different between groups at follow-up.
Conclusions:
GBA status appears to be an important predictor for non-motor symptom disease progression, after deep brain stimulation surgery.
INTRODUCTION
Parkinson’s disease (PD) is characterised clinically by motor symptoms [1], however, a host of non-motor symptoms also commonly occur. These include olfactory dysfunction, cognitive impairment, autonomic dysfunction, psychiatric symptoms, sleep disturbance, pain, and fatigue [2]. These symptoms can be very disabling and are associated with reduced health-related quality of life [3, 4].
While Levodopa is the most effective treatment for PD, it has significant limitations [5] including motor and non-motor fluctuations, dyskinesia, and psychosis [6]. Deep brain stimulation (DBS) surgery is a well-established treatment for the motor symptoms of PD, which involves the implantation of electrodes into the bilateral subthalamic nuclei (STN) or the globi pallidii interni (GPi) [7]. How DBS exerts its action is still not clear [8], however, in cases of DBS in the STN, its effectiveness allows for dopaminergic medication to be reduced post-operatively, which most certainly contributes to the reduction in drug-induced dyskinesias reported after surgery. DBS has been reported to significantly improve quality of life but is not a suitable option for everyone with PD. The contraindications to its usage include the presence of clinically relevant speech difficulties and cognitive impairment. These features may be exacerbated after DBS [9].
The clinical presentation of patients with PD is highly variable, which likely reflects a complicated interplay of environmental and genetic factors. Mutations in the glucosidase beta acid (GBA) gene are the most common genetic risk factor for developing PD [10] and have been associated with a more aggressive disease course. Evidence emerging over the preceding decade suggests GBA mutations predispose carriers to an earlier age of onset [10–13], accelerated progression of motor impairment [14–17], a greater burden of non-motor symptoms [18], and reduced quality of life [19]. Reports suggest that GBA associated PD carries a greater risk of cognitive impairment compared to idiopathic PD [10, 18–24]. However, the evidence reporting the clinical correlates of GBA-associated PD are based on a small number of patients and there are some uncorroborated and conflicting findings [25–27]. Further evidence is therefore required to elucidate the clinical features associated with GBA-associatedPD.
The aim of this study was to investigate the clinical features of PD patients, with and without GBA mutations, after deep brain stimulation surgery, exploiting the regular and detailed assessments that take place in this cohort of patients both before and after surgery, to explore whether outcomes differ according to GBA mutation status.
MATERIALS AND METHODS
Study design
This is a case-control study following 34 PD patients, with and without GBA mutations, after DBS surgery. Preoperative data were collected for each patient without knowledge of GBA status. Postoperative clinical assessments were completed in a non-blinded fashion.
Participants
All patients underwent DBS surgery between 2001 and 2011 at the National Hospital for Neurology and Neurosurgery (NHNN), London. Patients had been diagnosed according to the UK Parkinson’sDisease Brain Bank criteria and selected for surgery by a movement disorder multi-disciplinary team, following a battery of clinical assessments [15, 28]. DBS surgery was performed using an MRI-guided and MRI-verified technique described by Foltynie et al. [29]. All patients provided consent to genetic testing and were screened for mutations in SNCA, PARKIN, PINK1, DJ-1, LRRK2 and GBA. Genetic testing was performed by the Neurogenetics Unit at the NHNN, between 2008 and 2011, as previously described [15]. From this cohort, 17 patients with GBA mutations were identified (GBA+) in addition to a further 17 patients (GBA–) who were selected according to matched sex and disease duration to the GBA+ group, by an individual (VL) who had no previous contact with them, or knowledge of any of their clinical details.
The GBA+group included 15 heterozygous mutation carriers, 1 homozygous carrier, and 1 compound heterozygote. Two patients also carried a mutation in another PD-associated gene; PARKIN or LRRK2. The 17 patients who did not carry a GBA mutation (GBA–) were also negative for mutations in the other genes tested. All patients had previously consented to genotyping for PD genetic risks (REC number 08/H0715/121).
Clinical assessment
Postoperative clinical evaluation involved the administration of four validated assessments: The Mattis Dementia Rating Scale (DRS-2) was used to assess cognitive function; the assessment consists of 36 tasks comprising five subscales that provide information on specific abilities: attention, initiation/perseveration, constructional ability, conceptualisation and memory. A total raw score ranging from 0 to 144 is based on performance in all five subscales, with higher scores conferring better cognitive function. Age-corrected scores (the Age-Corrected Mayo’s Older Americans Normative Studies (MOANS) Scaled Score; AMSS) were obtained from the raw total score and each subscale score [30].
The severity of non-motor symptoms was assessed using the Non-Motor Symptom Assessment Scale for PD (NMSS); the scale comprises 30 items, each relating to a specific non-motor symptom. The items are classified in nine domains: cardiovascular, sleep/fatigue, mood/cognition, perceptual problems/hallucinations, attention/memory, gastrointestinal tract, urinary, sexual function and miscellaneous [31]. Domain scores are computed by the summation of the corresponding item scores, and for the total NMSS score by the sum of thedomains [32].
Motor symptoms were evaluated using part III of the Movement Disorder Society (MDS)-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS-III). Subscores for cardinal motor symptoms were derived by summation of the relevant items from the MDS-UPDRS as follows: axial symptoms (for example, speech and postural instability) (items 3.1, 3.2, 3.9–3.14), rigidity (item 3.3), bradykinesia (items 3.4–3.8) and tremor (items 3.15–3.18). Quality of life was measured using the Parkinson’s Disease Questionnaire(PDQ-39).
Participants were administered all assessments on a single day, which were completed in the same order. Each patient’s medication was recorded and their levodopa equivalent dose (LED) was calculated using a standard formula [33]. Stimulation settings were also documented during this time. All clinical assessments were performed with patients on-medication and on-stimulation.
Preoperative data, collected during the screening process for DBS surgery, were retrieved.
Statistical analysis
Mean scores and standard deviations were calculated for all continuous data and percentages were calculated for categorical values. Continuous data were checked for normality using the Shapiro-Wilk test (p = 0.05). If normally distributed, the unpaired Student’s t-test was used to compare GBA+ patients and GBA– patients. Alternatively, the Mann-Whitney U test was performed for non-parametric data. Categorical data was compared between groups using the Pearson’s chi-squared test, if the sample size was sufficient, or the Fisher’s exact test. All reportedp values are two-sided at a significance level of 0.05. Statistical tests were performed using the Stata 12.1 statistical software package (StataCorp LC, College Station, TX).
RESULTS
Demographics
Demographic characteristics of the 34 participants followed in this study are presented in Table 1.
Demographic characteristics of GBA+ and GBA- patients
aContinuous values are mean±standard deviation unless otherwise stated. bData available for 10 GBA+ patients and 16 GBA- patients. Abbreviations: n = number of patients; DBS = deep brain stimulation; STN = subthalamic nucleus. GPi = globus pallidus internus; LED = levodopa equivalent dose.
Despite being matched by disease duration, the mean age at surgery was 4 years younger in GBA+ patients, compared to non-carriers (t = –2.7, p = 0.015). Two GBA+ patients were selected for GPi DBS (based on the severity of dyskinesias) and all others underwent STN DBS. The mean time since DBS surgery was 7.5±2.0 years (range, 3.4 to 11.8 years) across all participants. At follow-up, 3 GBA+ patients (18%) and 1 GBA- patient (6%) were deceased. Furthermore, 2 GBA+ patients (12%) were unable to complete follow-up assessment due to severe PD-related disability and 2 could not be contacted (see Fig. 1). The remaining 26 patients completed follow-up clinical assessment (10 GBA+; 16 GBA-). There were no significant differences in the stimulation settings between GBA+ patients and GBA- patients at follow-up. However, the mean stimulation amplitude was higher in GBA+ patients (3.8±0.8 V) compared to GBA- patients (3.1±0.8 V). Pulse width was also slightly higher in the GBA+ group (66.0±10.5 μs vs 63.8±8.7 μs). Conversely, stimulation frequency was higher in GBA- patients (114.7±31.3 Hz vs 102.5±37.1 Hz).
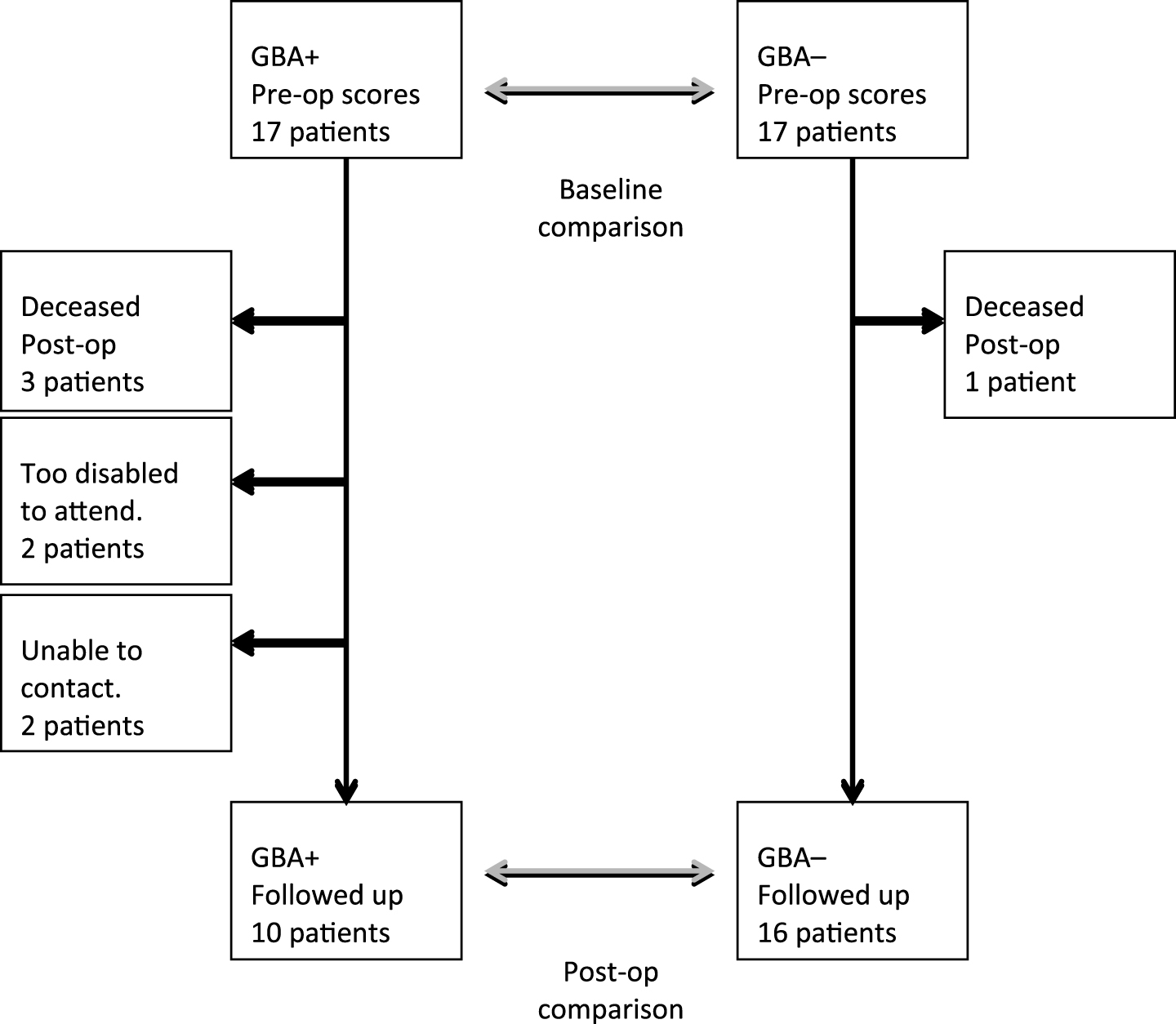
Flowchart of patients. GBA+ and GBA – patients were matched at baseline for sex and disease duration, by an individual blind to subsequent outcome.
Cognitive scores
At follow-up, the cognitive performance of GBA+ patients was significantly worse than GBA- patients (t = –3.1, p = 0.006; Table 2). The mean AMSS scores of GBA mutation carriers were consistently lower across all five cognitive domains. Seven of 10 (70%) GBA+ patients were cognitively impaired compared to 3 of 16 (19%) GBA- patients (χ2 = 6.8, p = 0.009). Moreover, cognitive impairment was more severe in GBA+ associated PD. Six (60%) cognitively impaired GBA+ patients were considered severely impaired. Among GBA- patients with cognitive impairment, only 1 (6%) demonstrated severe cognitive impairment.
Cognitive performance of GBA+ patients and GBA- patients
Values are mean±standard deviation unless otherwise stated. All scores are on a scale of 2 to 18. Higher scores denote better cognitive performance. aData available for 10 GBA+ patients and 6 GBA- patients. Abbreviations: n = number of patients; DRS-2 = Mattis Dementia Rating Scale; AMSS = Age-corrected Mayo’s Older Americans Normative Studies (MOANS) Scaled Score.
According to clinical guidelines used to select candidates for DBS, all patients were cognitively intact preoperatively; although 2 patients performed below average (1 GBA+; 1 GBA-). There was no significant difference in pre-operative cognitive performance, however, due to the variability in the neuropsychological tools used preoperatively, DRS-2 scores were available for only 16 patients.
Non-motor scores
The mean total NMSS score was significantly higher for GBA+ compared to GBA- (t = 3.2, p = 0.004; Table 3) indicating a greater burden of non-motor symptoms in this group. The mean scores of the GBA+ group were higher across all domains, except cardiovascular symptoms, which was marginally lower. Notably, the mean score for attention/memory in GBA+ patients was more than double that of GBA- patients. Preoperative NMSS scores were not available for the study cohort, as it was not routinely administered prior to DBS at the time of theirsurgery.
Non-motor performance of GBA+ patients and GBA- patients
Values are mean±standard deviation unless otherwise stated. Higher scores denote greater non-motor symptom burden. Abbreviations: n = number of patients; NMSS, Non-motor symptom assessment scale for Parkinson’s disease.
Quality of life scores
Compared to GBA- patients at follow-up, GBA+ patients reported significantly worse quality of life (t = 2.2, p = 0.040). The GBA+ group demonstrated greatest deterioration in mobility, activities of daily living, cognition, and communication (Table 4). The quality of life of patients, with and without GBA mutations, did not differ preoperatively.
Quality of life scores of GBA+ patients and GBA- patients
Values are mean±standard deviation unless otherwise stated. All scores are on a scale of 0 to 100. Higher scores denote reduced quality of life. Abbreviations: n = number of patients; PDQ-39 = Parkinson’s Disease Questionnaire; SI = summary index; ADL = activities of daily living.
Motor scores
Preoperative, on-medication motor scores did not differ significantly between patients with GBA mutations and those without mutations (Table 5). However, off-medication, preoperative total UPDRS-III scores were significantly higher in the GBA+ group (t = 2.7, p = 0.009)), indicating greater motor impairment in this group. Analysis of UPDRS-III scores show that rigidity and axial symptoms were significantly worse in the GBA+ group (t = 2.6, p = 0.015 and t = 2.3, p = 0.026, respectively). Tremor and bradykinesia were not significantly different between GBA+ and GBA- groups.
Motor performance of GBA+ patients and GBA- patients
Values are mean±standard deviation unless otherwise stated. Higher scores denote greater disability. aData available for 9 GBA+ patients and 16 GBA- patients; 1 GBA+ patient did not complete motor assessment due to having his DBS hardware removed in May 2016, following erosion of the hardware. Abbreviations: n = number of patients; UPDRS-III = Part III of the Unified Parkinson’s Disease Rating Scale; MDS-UPDRS-III = Part III of the Movement Disorder Society-sponsored Unified Parkinson’s Disease Rating Scale.
At follow-up, the on-medication MDS-UPDRS-III scores were higher among GBA+ patients with accompanying higher motor scores in all domains except tremor, however, none of these motorcomparisons achieved conventional thresholds for statistical significance.
DISCUSSION
Clinical features of GBA-associated PD after deep brain stimulation (DBS)
The results of this study are consistent with previous reports, that GBA status influences the natural progression of disease, in a DBS cohort. Although age at onset, preoperative cognition, and quality of life were similar between PD patients with and without GBA mutations, GBA+ patients required DBS earlier in their disease course, suggesting a faster rate of progression. This is also reflected in their preoperative, off-medication motor performance. The observation that 5 GBA+ patients could not participate in follow-up assessment, due to severe disability or death, supports this suggestion. An increased rate of mortality has been demonstrated in GBA mutation carriers [17]. Importantly, the loss of these 5 patients from follow-up represents a bias, as those who had/have the greatest disease severity could not be included in follow-up analysis. As a consequence, the differences found between GBA+ and GBA- patients at follow-up are likely to be underestimates.
Follow-up data revealed cognitive impairment to be more prevalent (70% vs 19%) and more severe (60% vs 6% were severely impaired) in GBA mutation carriers, corroborating a growing body of evidence that find GBA mutations increase the risk and rate of cognitive decline. These findings correlate with pathological reports of a higher density of Lewy pathology in the neocortex of these patients [13, 35]. The DRS-2 performance of those with GBA+ associated PD conformed to the expected pattern of PD, and mimicked the pattern of non-carriers, with lowest scores in the construction and initiation/perseveration domains [36]. This indicates that GBA status does not alter the form of cognitiveimpairment but influences the rate of cognitive decline. This contrasts with the findings of a large, multicentre study that found GBA mutations to be associated with a distinct cognitive profile characterised by greater impairment in working memory, executive function and visuospatial abilities in PD patients [37].
GBA mutation carriers exhibited a greater burden of non-motor symptoms at follow-up. The prevalence and spectrum of non-motor symptoms appeared indistinguishable between groups, but the severity of symptoms was much worse in GBA-associated PD; particularly sleep/fatigue, attention/memory, gastrointestinal, and urinary symptoms.
At a mean follow-up of 7.5 years, the quality of life of GBA- patients had almost returned to the preoperative status, after a period of effective DBS therapy [15]. Although there are very few reports of long-term outcomes after DBS, this finding is consistent with a review, which found quality of life in PD patients significantly improved 1 year post-DBS and back to baseline by 5 years follow-up [38]. For GBA+ patients, the mean quality of life score was, however, significantly worse at follow-up. This finding corroborates the results of a previous study which investigated the impact of GBA status on quality of life, irrespective of DBS [19]. According toprevious reports, it can be inferred that the reduced quality of life observed in GBA mutation carriers is directly related to their increased burden of non-motor symptoms [3, 4]. It must be noted that the mean ‘emotional’ score of GBA+ patients was slightly higher than GBA- patients, denoting lower mood. Depression has been reported one of the main determinants of quality of life in PD and it is plausible that a low mood would impact on an individual’s self-assessment of most domains of quality oflife [39].
Given the accumulating evidence implicating GBA mutations with a greater severity of non-motor symptoms, it has been suggested that GBA mutations predispose carriers to a more aggressive disease. The mean total MDS-UPDRS-III score was 11 points higher in GBA mutation carriers, accompanied by higher stimulation settings and higher LED requirements. There are a number of small studies that have suggested GBA mutations increase the severity of motor symptoms. While this may be evident when assessed off-medication or in the later aspects of disease, it appears likely that patients are responsive to dopamine replacement based on our pre-operative comparisons which are consistent with those of a recent meta-analysis which found no statistical difference between the UPDRS-III scores of 646 GBA+ patients and 10,344 GBA- in the on-medicationstate [27].
Clinical relevance
Determining the effects of genetic mutations on PD phenotype is important to improve our ability to counsel PD patients as to their likely disease course. In GBA+ associated PD, the reported incidence of increased burden of non-motor symptoms, particularly the increased likelihood and accelerated cognitive decline, can be a great source of concern for patients and their families. A personalised medicine approach involves providing patients with greater certainty about their individual prognosis and enhancing their ability to make informed decisions about therapeutic choices.
Notably, not all GBA mutation carriers demonstrate the clinical features associated with more aggressive disease. In this report, 30% of GBA mutation carriers who completed the DRS-2 at follow-up were cognitively intact. Similarly, some individuals with GBA-associated PD reported less severe non-motor symptoms and better quality of life than some non-carriers. Although these individuals are exceptions to the emerging trend observed in GBA+ associated PD, it is clear that there are other factors determining the clinical outcomes of PD patients and further research is required to understand what these additional modifiers are. As the ultimate aim of DBS for PD is to improve the quality of life of patients, results presented here suggest that GBA status may be an important consideration when weighing the benefits and major risks associated with the surgery. According to their PDQ-39 follow-up assessment, by a mean time of 8.6 years post-operatively, GBA mutation carriers’ quality of life becomes worse compared to their preoperative scores. One could speculate that these patients are less responsive to the effects of DBS or perhaps only benefit for a short time period, presumably due to more rapid progression of PD. Whether the decline observed is the result of GBA mutations alone or because of an interaction between GBA status and DBS is not known. These results might, however, suggest the use of bilateral GPi-DBS, rather than bilateral STN-DBS, may be favourable because additional negative cognitive consequences, such as diminished verbal fluency, have been reported in STN-DBS [40–42].
There are several limitations that should be taken into consideration when reviewing the findings of this study. GBA mutation carriers were matched for sex and disease duration, as both motor and non-motor symptoms are primarily influenced by the duration of symptoms. Nevertheless, other variables may have influenced the outcome measures. This study explores phenotypic differences in a DBS cohort who were selected as appropriate candidates during pre-operative screening. Accordingly, the results presented here should be considered in this context, as data may not be representative of the wider PD population. Individuals who undergo DBS tend to have a young age at onset, which holds true in the study cohort who had a mean age at onset of 42 years. Additionally, to be eligible for surgery, patients must not have exhibited clinically relevant cognitive impairment or neuropsychiatric disturbance.
The greatest limitation of the study is sample size. The relatively small number of patients in each group limits the power of the study to reach definite conclusions. Furthermore, despite our best efforts at matching the 2 groups, there are small but non-significant differences between the groups in terms of age and disease duration that may partly contribute to the different rates of progression that we have observed. Nevertheless, the magnitude of the differences that emerge despite the small sample size are consistent with the emerging view that GBA+ patients have a more aggressive form of PD.
The findings presented here require validation in larger patient cohorts to explore the relevance of different GBA mutations on phenotype and investigate whether zygosity influences the clinical presentation of carriers. Furthermore, it would be of interest to investigate the clinical outcomes of GBA mutation carriers, according to DBS target. In conclusion, GBA status appears to be an important predictor for non-motor symptom disease progression in PD. These results associate GBA mutations with more prevalent and more severe cognitive impairment and a greater burden of non-motor symptoms. Together, these findings suggest that GBA mutations predispose carriers to a more aggressive form of PD.
CONFLICT OF INTEREST
TF, MH, PL, LZ and JHyam have received honoraria for speaking at meetings sponsored by Medtronic. None of the other authors have any conflicts of interest relevant to this study.
Footnotes
ACKNOWLEDGMENTS
We would like to thank all the patients who participated in this study and their families.
This research was supported by a grant from Parkinson’s UK (K-0901). This work was undertaken partly at UCL/UCLH and was funded in part by the Department of Health NIHR Biomedical Research Centres funding scheme. The Unit of Functional Neurosurgery, UCL Institute of Neurology, Queen Square, London, is supported by the Parkinson’s Appeal and the Monument Trust.