
Abstract
Chronic progressive external ophthalmoplegia (CPEO) is symptom complex with progressive ptosis and restricted ocular motility without diplopia. MYH2 myopathy is rare disorder presenting with CPEO and muscle weakness. We report two Indian patients of MYH2 myopathy with unique features. Patient-1 presented with early adult-onset esophageal reflux followed by, proximal lower limb weakness, proptosis, CPEO without ptosis. He had elevated creatine kinase along with characteristic muscle MRI findings of prominent semitendinosus and medial gastrocnemius involvement. Patient -2 presented with early adult onset CPEO without limb weakness. His creatine kinase was normal. Both the patients had novel MYH2 mutations: a homozygous 5’splice variation in intron 4 (c.348 + 2dup) in patient 1 and homozygous single base pair deletion in exon 32 (p. Ala1480ProfsTer11) in patient 2. Unique features noted include adult onset, isolated CPEO, proptosis, esophageal reflux disease and absence of skeletal abnormalities. MYH2 myopathy has to be considered in adult patients with CPEO.
INTRODUCTION
Chronic progressive external ophthalmoplegia (CPEO) is a clinical syndrome characterized by symmetric bilateral ptosis with restricted ocular motility. CPEO is seen in various neuromuscular conditions such as mitochondrial disorders, centronuclear myopathies, congenital myasthenic syndromes and oculopharyngeal muscular dystrophy. MYH2 (myosin heavy chain-2) related myopathies are a group of congenital myopathies which present with ptosis and external ophthalmoplegia [1]. They can present as autosomal dominant (AD) mutation with congenital joint contractures, CPEO and proximal muscle weakness [2]. Autosomal recessive (AR) mutations in MYH2 also present with external ophthalmoplegia with proximal weakness but is largely mild and non-progressive [3].
Here we present two interesting cases of homozygous MYH2 myopathy with CPEO and progressive muscle weakness.
This was a retrospective review on two unrelated patients with genetically proven MYH2 myopathy from a neuromuscular division of a quaternary Neurology centre in India. Detailed clinical, laboratory and electrophysiological data was recorded and is being presented. The two patients provided written informed consent for publication of their case details, the investigation results and clinical pictures with unmasked faces. Patient-1 underwent MRI muscle at MRI 1.5 T [Aera; Siemens Healthcare; Erlangen, Germany with body coil at 3 stations - Hip to Upper Thigh, Upper Thigh to Knee, Leg, FOV (Field Of View). Muscle analysis involved visual assessment for fatty infiltration and muscle volume loss [4].
CASE REPORT
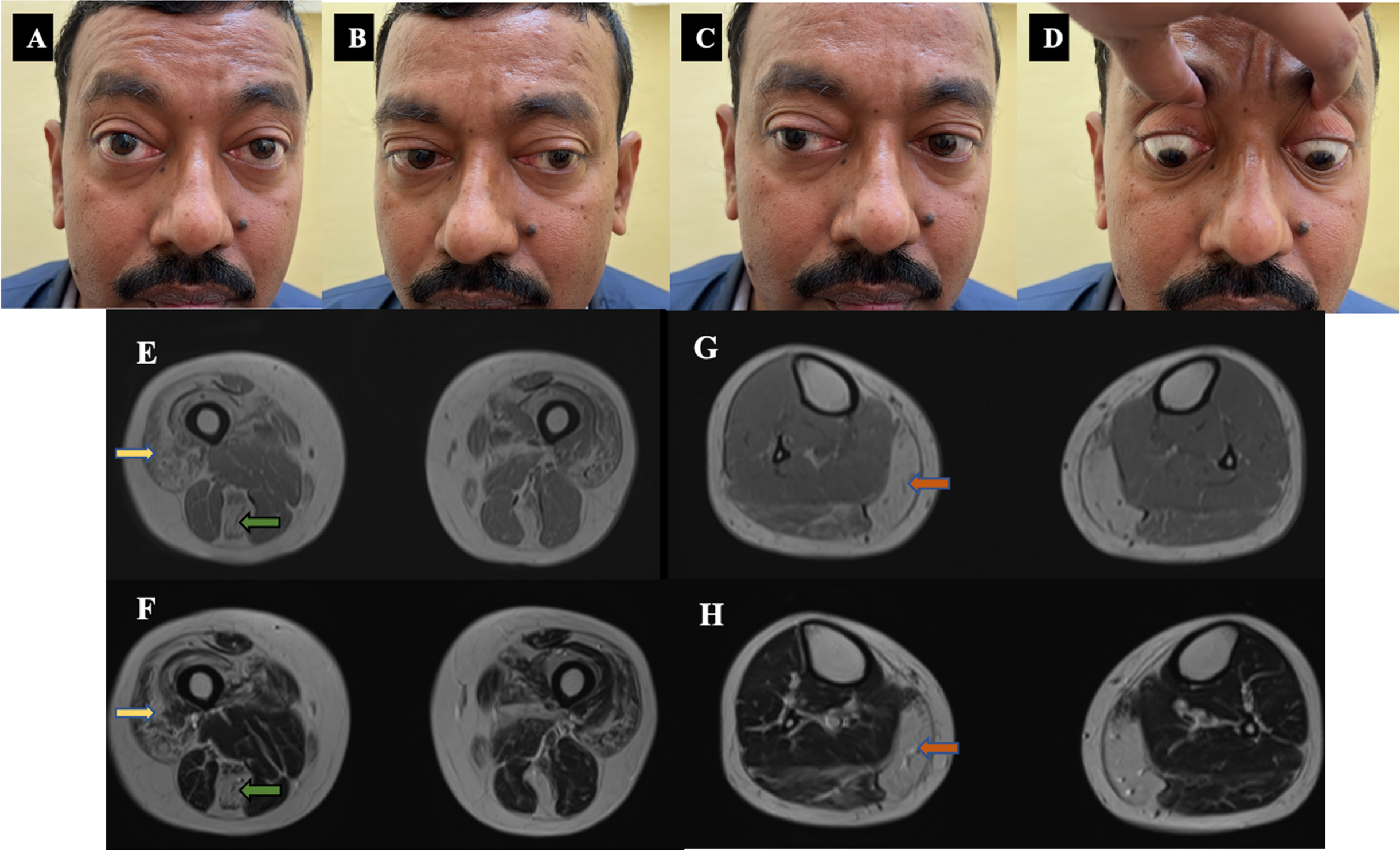
Clinical images and MRI muscle of patient –1. A, B, C, D: Clinical images - Ocular motility restriction - Abduction and upgaze restriction with proptosis. E, F, G, H: Muscle MRI images - E- T1 W, F-T2 W images showing quadriceps (yellow arrow), semitendinosus (green arrow) fatty infiltration. G-T1 W, H-T2 W images showing predominant medial gastrocnemius (orange arrow) fatty infiltration with sparing of anterior leg muscles.

A, B, C, D: Clinical images of patient-2. A –Asymmetric ptosis. B, C- Restricted ocular motility. D,E –Facial weakness.
GENETIC RESULTS
Clinical exome sequencing was performed using custom capture kit in both patients revealing rare homozygous MYH2 variants: In patient 1, a novel single nucleotide duplication c.348 + 2dup (hg38; chr17: g.10547473dupA) at 5’ splice site in intron 4 was identified which was predicted as splice-altering (0.5 score) by splice AI tool [5]. In patient 2, a single nucleotide deletion c.4438del (hg38; chr17: g.10525550del) resulting in frameshift (p. Ala1480ProfsTer11) was identified which was previously reported as pathogenic in clinvar [6]. Both variants are absent in population databases including gnomAD and GenomeAsia 100K as well as internal databases [7]. Based on this evidence both variants are considered as putative disease-causing although further functional validation and segregation in unaffected family members is not performed. Mitochondrial genome sequencing was negative in both patients.
DISCUSSION
Myosinopathies are a group of ultra-rare inherited muscle disorders with varied phenotypic features involving mutation in myosin heavy chain of skeletal muscle (MYH) genes in chromosome 17 [8]. MYH2 is one of the three isoforms of myosin heavy chains expressed mainly in fast twitch 2A and 2B fibres. AD-MYH2 myopathy usually presents with transient congenital arthrogryposis followed by late onset progressive proximal limb weakness and external ophthalmoplegia. It has characteristic muscle biopsy features of reduced type 2A fibres and rimmed vacuoles [9]. AR-MYH2 myopathy is characterised by mild early onset non-progressive proximo-distal weakness of limbs with external ophthalmoparesis with biopsy features of complete absence of type 2A fibres and non-specific myopathic changes [8]. Our patients presented with AR-MYH2 myopathy with unique features in contrast to previously described phenotypic features. Both the patients had adult onset of symptoms. Isolated fatigable CPEO was noted in patient -2 without limb weakness mimicking mitochondrial myopathy and congenital myasthenic syndromes. The prominent ptosis noted in patient - 2 is also rarely described in AR-MYH2 myopathy [8]. Proptosis in our patient (Patient-1) has not been described previously. The prominent postprandial esophageal reflux was noted in patient-1 since early adulthood. This is possibly due to the skeletal muscle MYH2 involvement of the upper esophagus resulting in supine gastric reflux which is a novel manifestation noted [10]. The muscle MRI done in patient -1 showed characteristic differential muscle involvement of antero-medial thigh and medial gastrocnemius. The predominant involvement of semitendinosus with relative sparing of other hamstring muscles has been consistently noted in previous reports which may be a pointer towards MYH2 myopathy diagnosis [1, 11]. Hence MRI muscle can be considered in patients with unexplained external ophthalmoparesis which can help in identifying these patients. Both the variants noted in our patients are novel mutations which may be responsible for the unusual presentations noted in our patients as described. However, functional studies were not performed.
Thus, MYH2 myopathy needs to be considered in the differential list in patients presenting with CPEO even in the absence of proximal myopathy. The absence of decremental response in RNS along with characteristic muscle MRI findings may aid in early diagnosis which can be confirmed with next generation sequencing-based genetic diagnosis.
Footnotes
ACKNOWLEDGMENTS
Nil.
SOURCES OF SUPPORT
Nil.
DISCLOSURE OF FUNDING
Nil.