
Abstract
Mitochondrial Calcium Uptake 1 (MICU1) is an important component of mitochondrial calcium channel regulator. Mutations in MICU1 result in a rare syndrome of myopathy with extrapyramidal features. Here we report a rare case of MICU1 related myopathy from India. A 23 years old male presented with 10 years history of proximal muscle weakness with exertional myalgia and fatigue. Examination showed facial dysmorphism with facial weakness and mildly reduced visual acuity. Limb girdle pattern of weakness with hypoactive tendon reflexes were noted without extrapyramidal signs. He had elevated serum creatine level of 1542 IU/L. Muscle MRI had novel findings of selective fatty infiltration of hamstrings, medial gastrocnemius and soleus. Muscle biopsy showed myopathic with secondary neurogenic changes along with few COX deficient fibres. Genetic analysis showed compound heterozygous pathogenic variants in MICU1 gene at intron 9 (c.1072-1 G > C) – splice site variant and exon 5 (c.513T > A) – stop gained variant, both resulting in loss of function of the protein. The variants were segregating in unaffected parents in heterozygous state with variant c.1072-1 G > C in the unaffected father and variant c.513T > A (p. Tyr171*) in the unaffected mother confirming the diagnosis. This report highlights the phenotype of limb girdle weakness with facial dysmorphism and optic atrophy expanding the spectrum of MICU1 related syndrome with novel MRI muscle and histopathological findings.
Introduction
Mitochondrial calcium (Ca2+) uptake is a key mediator of cell survival, metabolism and apoptosis which is tightly regulated. This also plays an important role in skeletal muscle contraction, energy metabolism and sarcolemmal repair apart from neuronal maintenance of brain. 1 Mitochondrial Calcium Uptake 1 (MICU1) is a subunit of Mitochondrial Calcium Uniporter (MCU) in the inner mitochondrial membrane and acts as a Ca2+ sensor. 2 The major function of MICU1 is setting the threshold of extramitochondrial Ca2+ load for mitochondrial Ca2+ uptake and protects mitochondria from Ca2+ overload. 3 The MICU1 gene is located on chromosome 10q22. Biallelic mutations in MICU-1 are associated with a rare syndrome of myopathy and extrapyramidal signs called myopathy with extrapyramidal signs (MPXPS). This is an ultra-rare disorder with less than 50 cases reported worldwide.4,5,6 MPXPS is a heterogenous disorder with varied manifestations even with patients having same pathogenic variants. The typical syndrome of MPXPS is characterised by proximal muscle weakness, cognitive disturbance, raised creatine kinase levels and extrapyramidal features. 7
Here we report a case of MICU-1 associated myopathy without extrapyramidal features from India. The details of clinical, radiological, genetic and pathological findings are being described. Institutional ethical approval to collect and data from the medical records was obtained. Magnetic Resonance Imaging (MRI) of Brain and lower limbs were done. Muscle biopsy was performed from biceps brachii muscle and was subjected to routine histopathological analysis including eosin and hematoxylin stain, modified Gomori's trichrome and COX staining. Genetic analysis was performed by exome sequencing as previously described. 8 Identified variants were classified as pathogenic/likely pathogenic as per American College of Medical Genetics (ACMG) criteria. 9 Informed consent was obtained from the patient for publication of clinical details and photographs.
Case report
Clinical details
A 23 years-old-male from southern part of India was evaluated during the year 2022. He presented with proximal limb weakness for 10 years. He had difficulty in rising from floor and climbing stairs along with difficulty in doing overhead activities. He also had exertion induced myalgia of legs with fatigue. There were no cerebellar or cranio-bulbar symptoms. He was born to non-consanguineous parents and had no significant family history. The developmental milestones were appropriate for age as per the parents and started to walk independently around 12–18months of age. On examination, he had a slender habitus and facial dysmorphism with low set ears, temporal hollowing and maxillary hypoplasia (Figure 1(A), (B) and (C)). He had flexion contractures of fingers and toes with mild facial weakness, scapular winging and exaggerated lumbar lordosis. Visual acuity was 6/9 bilaterally. Fundus examination showed bilateral temporal pallor. According to Medical Research Council (MRC) grading, the muscle power of neck flexor was grade 2, deltoid and pectoralis major (5), biceps brachii (3), triceps (5), wrist (5), iliopsoas and gluteus maximus (3), hip abductors (4), hip adductors (3), quadriceps femoris (4), hamstrings (3) and ankle (4). The tendon reflexes were hypoactive. The cerebellar and extrapyramidal systems were normal. He had a waddling gait with toe walking. The possibilities of limb girdle syndrome with facial dysmorphism such as congenital myopathy and mitochondrial myopathy were considered.
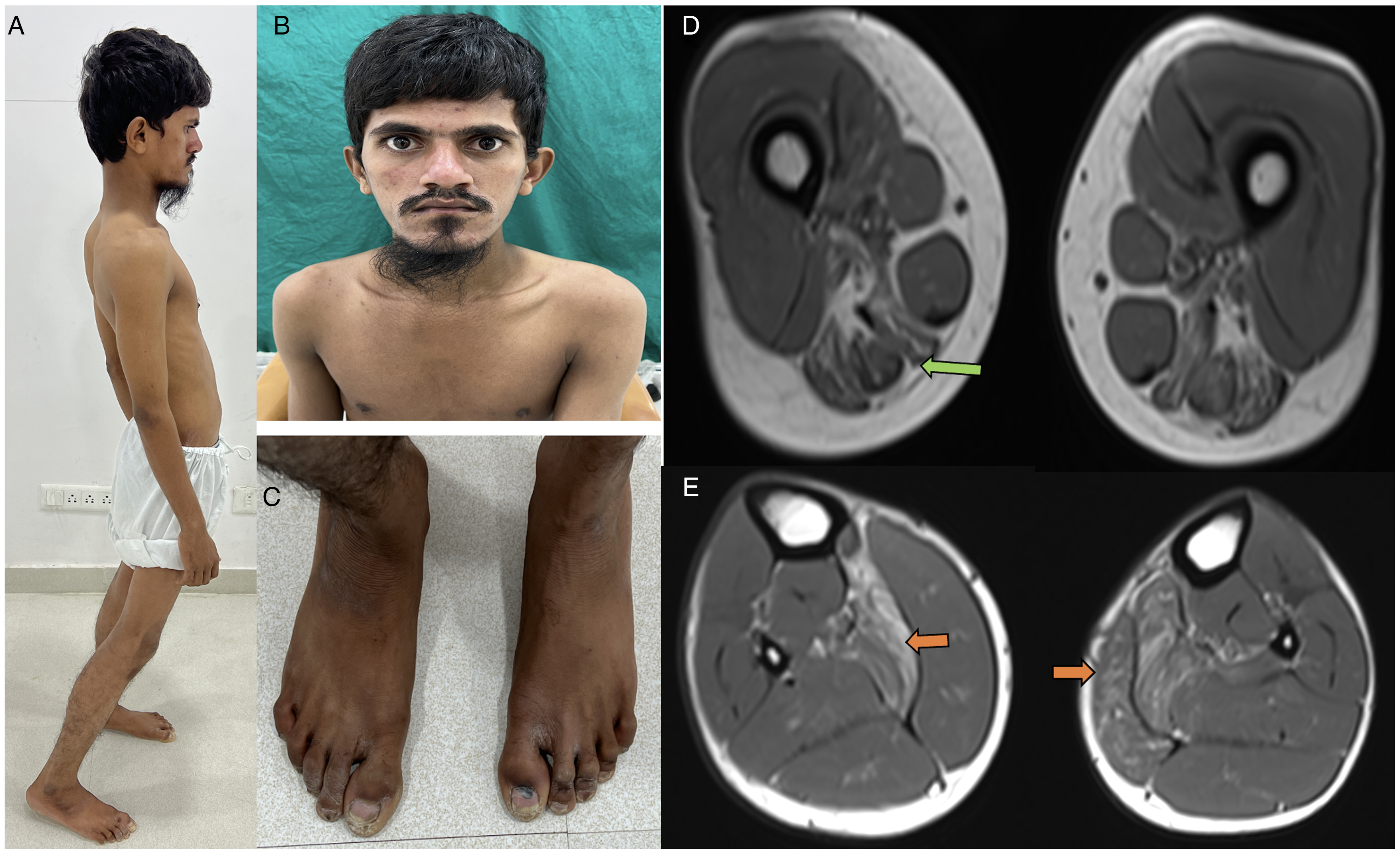
Clinical images and MRI muscle of the patient:
Laboratory investigations
Investigations showed elevated creatine kinase levels of 1542 IU/L. The blood lactate, thyroid profile, calcium and phosphorus and liver enzymes were within normal limits. Nerve conduction study and repetitive nerve stimulation revealed no abnormality. Visual evoked potentials showed prolonged P-100 latencies (right: 109 ms, left: 112.5 ms). Somatosensory potentials and auditory evoked responses were normal. Muscle magnetic resonance imaging (MRI) showed fatty infiltration in hamstrings, medial gastrocnemius and soleus (Figure 1(D) and (E)). MRI brain was normal. Muscle biopsy from biceps brachii showed mild myopathic with secondary neurogenic changes with occasional COX deficient fibres (Figure 2). Mitochondrial assays were normal.
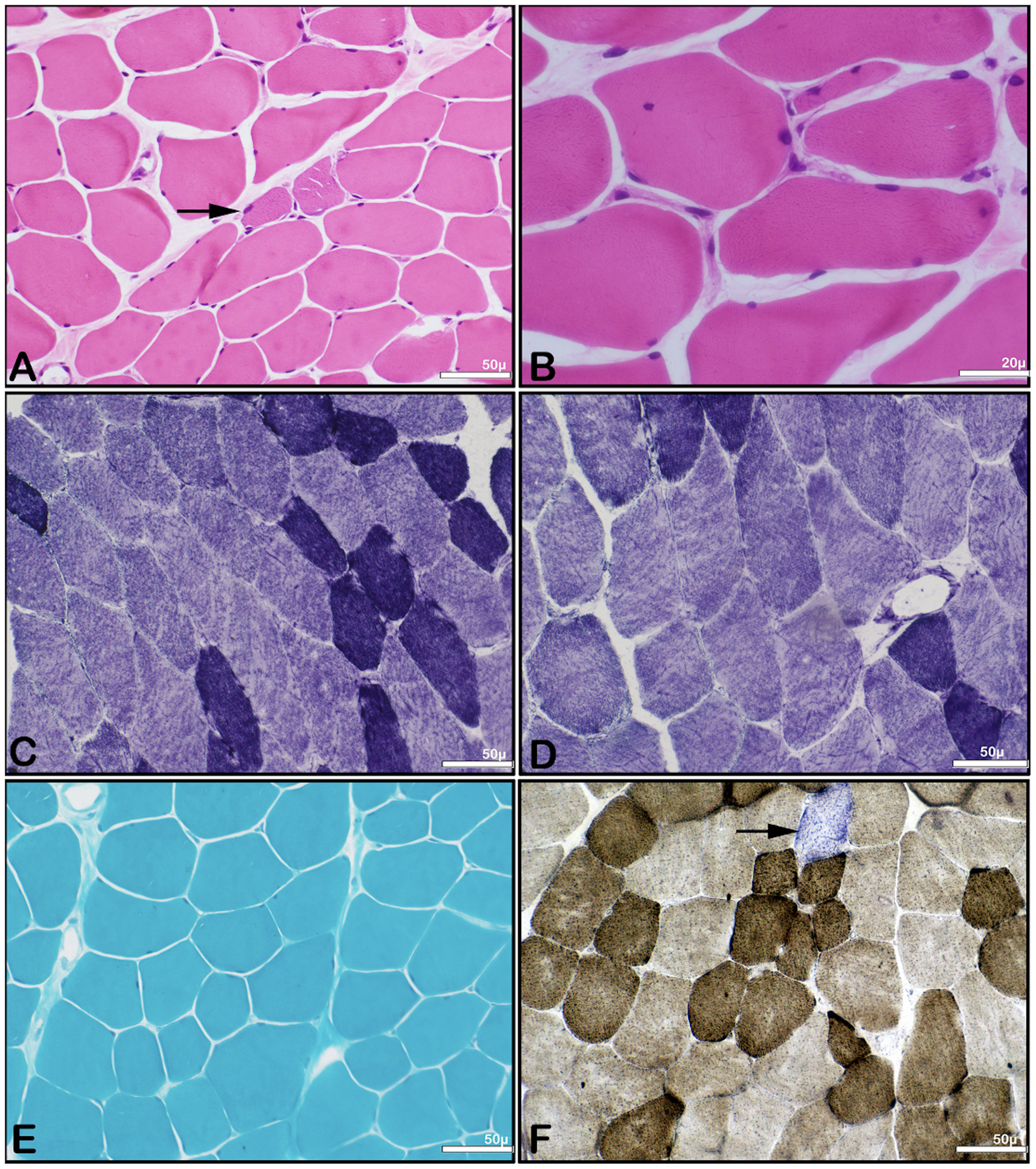
Histopathology images of muscle biopsy of the patient:
Genetic analysis
Clinical exome sequencing and mitochondrial genetics were done (LOVD Submission Number: 00445138) which showed pathogenic compound heterozygous variants in MICU1 gene involving intron 9 - c.1072-1 G > C (NM_001195518.2) and exon 5 - c.513T > A, p.Tyr171Ter (NM_001195518.2) of GRCh38: chr10:72533770 A > T.
Variant c.1072-1G > C
The variant c.1072-1G > C is classified as pathogenic (PVS1, PP5, PP4).
PVS1: This variant results in the loss of an acceptor splice site for the clinically relevant transcript. This variant also disrupts the acceptor splice site for an exon upstream from the last coding exon resulting in a frameshift variation that is predicted to cause nonsense mediated decay. The c.1072-1G > C variant is a loss of function variant in the gene MICU1, which is a known mechanism of disease, as indicated by the presence of existing pathogenic loss of function variant NP_001182447.1:p.M1V and 16 others.
PP5: The splice acceptor variant NM_001195518.2: c.1072-1G > C has been reported to ClinVar as Pathogenic with a status of (1 star) criteria provided, single submitter (Variation ID 101045 as of 2023-03-02). The variant is present in 35/ 1455458 (AF = 0.00002405) alleles from individuals in gnomAD Exomes database only in heterozygous state. The variant is present in 1/ 152170 (AF = 0.000006572) alleles from individuals in gnomAD Genomes database only in heterozygous state. This variant mutates a splice-acceptor sequence and is predicted to disrupt the reading frame, resulting in nonsense mediated decay. This variant results in the loss of an acceptor splice site for the clinically relevant transcript. This variant disrupts the acceptor splice site for an exon upstream from the last coding exon resulting in a frameshift mutation that is predicted to cause nonsense mediated decay. The c.1072-1G > C variant is a loss of function variant in the gene MICU1, which is a known mechanism of disease, as indicated by the presence of existing pathogenic loss of function variant NP_006068.2:p.M1V and 17 others.
PP4: Patient's phenotype or family history is highly specific for a disease with a single genetic etiology.
In addition, the clinical phenotype of the proband partially matches with that of the disorder caused by pathogenic variants in the MICU1 gene. For these reasons, this variant has been classified as Pathogenic.
Variant c.513T > A (p.Tyr171Ter)
The variant c.513T > A (p.Tyr171Ter) is classified as Pathogenic (PM2 PVS1 PP4).
PM2: The stop gained NM_001195518.2: c.513T > A variant is novel (not in any individuals) in 1kG All and gnomAD as well as in our inhouse database. The variant has not been reported previously as a pathogenic variant nor as a benign variant, to our knowledge.
PVS1: This variant is a stop gained variant which occurs in an exon of MICU1 gene upstream of where nonsense mediated decay is predicted to occur. There are 14 downstream pathogenic loss of function variants, with the furthest variant being 179 residues downstream of this variant. This indicates that the region is critical to protein function. The p.Tyr171Ter variant is a loss of function variant in the gene MICU1, which is a known mechanism of disease, as indicated by the presence of existing pathogenic loss of function variant NP_006068.2:p.M1V and 17 others. In addition, the clinical phenotype of the proband partially matches with that of the disorder caused by pathogenic variants in the MICU1 gene.
PP4: Patient's phenotype or family history is highly specific for a disease with a single genetic etiology. For these reasons, this variant has been classified as Pathogenic.
Also, the pathogenic variants in MICU1 gene were segregating in unaffected parents in heterozygous state with variant c.1072-1 G > C in the unaffected father and variant c.513T > A (p. Tyr171*) in the unaffected mother. Hence, the diagnosis of MICU-1 related myopathy was confirmed in the proband.
Discussion
Our patient presented with slowly progressive limb girdle pattern of weakness with exertion induced fatigue, facial dysmorphism and optic atrophy without extrapyramidal features. Previous large cohorts are reported in Middle eastern and UK-Pakistani populations.7,10,11 In the Middle Eastern cohort, extrapyramidal features were noted in 35% and proximal muscle weakness in 52.6% of patients. Facial dysmorphisms, developmental delay and speech abnormalities were other features seen in this group.10,11 Another cohort of UK-Pakistani patients by Logan et al., presented mainly with extrapyramidal features and speech disturbances (72.7%). 7 None of these patients had isolated myopathy. The presence of exertion induced myalgia and fatigue noted in our patient was previously reported by Lewis et al., in a patient from United Kingdom. 10 These patients had homozygous deletion of exon 1 of MICU1 within a 2755-base pair deletion. The fatigue may be due to mitochondrial Ca2 + channelopathy. 12 Muscle biopsies done in MICU1 related syndromes showed non-specific myopathic changes.7,13 However, another report by Roos et al., showed myopathic changes with secondary neurogenic changes. 14 None of the previous reports showed mitochondrial features of COX deficient fibres as noted in the current report. Previous reported Indian report of MICU1 related syndrome showed MRI features of fatty infiltration of adductors and gluteus maximus.5 However, our patient showed notable involvement of posterior compartment of thighs and legs which may be related to the novel pathogenic variant in the current report. The most common variants reported are homozygous pathogenic variants with few compound heterozygous pathogenic variants. 4 The most common variant is c.533C > T reported in twenty middle eastern Arab patients predominantly in homozygous state.10,11 Logan CV et al., reported the pathogenic splice site variant c.1078-1G > C (with reference to NM_006077.4 transcript) as noted in the current study (c.1072-1G > C with reference to NM_001195518.2 transcript) but in homozygous state. 7 Our report also has shown a novel stop gained pathogenic variant in c.513T > A in exon 5 of MICU1 gene.
This report highlights recognition of this rare slowly progressive myopathy with fatigue, facial dysmorphism and optic atrophy. Important novel MRI muscle and histopathological features have also been noted. MICU1 related myopathy has to be considered in the differential diagnosis of suspected mitochondrial myopathies.
Comparison with previous studies is shown in Supplementary Table.
Supplemental Material
sj-docx-1-jnd-10.1177_22143602241288400 - Supplemental material for MICU1 related myopathy – a rare report from India
Supplemental material, sj-docx-1-jnd-10.1177_22143602241288400 for MICU1 related myopathy – a rare report from India by Dipti Baskar, Suma Reddy Ganji, Aneesha Thomas, Kiran Polavarapu, Bevinahalli N. Nandeesh, Sai Bhargava Sanka, Kosha Srivastava, Ananthapadmanabha Kotambail, Gautham Arunachal, Vijay Kumar Boddu, Saraswati Nashi, Atchayaram Nalini and Seena Vengalil in Journal of Neuromuscular Diseases
Footnotes
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.