
Abstract
We describe a patient with chronic progressive external ophthalmoplegia (CPEO) due to a rare mitochondrial genetic variant. Muscle biopsy revealed numerous cytochrome c oxidase (COX)-deficient fibres, prompting sequencing of the entire mitochondrial genome in muscle which revealed a rare m.12334G>A variant in the mitochondrial (mt-) tRNALeu(CUN)(MT-TL2) gene. Analysis of several tissues showed this to be a de novo mutational event. Single fibre studies confirmed the segregation of high m.12334G>A heteroplasmy levels with the COX histochemical defect, confirming pathogenicity of the m.12334G>A MT-TL2 variant. This case illustrates the importance of pursuing molecular genetic analysis in clinically-affected tissues when mitochondrial disease is suspected.
INTRODUCTION
Mitochondrial DNA point mutations are a significant contributor to human disease with some population studies demonstrating a prevalence of greater than one in 200 live births [1]. They are associated with variable clinical phenotypes and can present in childhood or adulthood [2]. In around 75% of cases, mutations are strictly maternally inherited [2]. Around 25% occur de novo [2]. Mitochondrial transfer RNA (mt-tRNA) genes constitute around 9% of the entire mitochondrial genome, have a crucial role in mitochondrial protein synthesis is and when mutated, can produce a wide diversity of clinical phenotypes [3]. In most cases, multiple highly oxidative tissues are affected resulting in multisystemic disease [4]. It is rare for mt-tRNA variants to present with isolated organ involvement; isolated CPEO with or without exercise intolerance is reported in only 4% of cases [5]. At least 40 mt-tRNA variants are associated with mitochondrial myopathy or PEO [4, 6–8].
CPEO can result from either primary or secondary mitochondrial DNA defects. Single, usually sporadic, large deletions of mitochondrial DNA occurring in early embryogenesis frequently cause CPEO [9]. CPEO is a hallmark of multiple mtDNA deletion disorders, characterized by a mosaic COX defect in clinically-relevant tissues with affected fibres containing high levels of a clonally-expanded mtDNA rearrangement. CPEO is also a consequence of point mutations in mitochondrial DNA, either maternally-inherited or arising de novo. Secondary mitochondrial defects causing CPEO result from autosomal dominant or recessive mutations in nuclear DNA in genes encoding proteins involved in mtDNA maintenance, replication or repair such as POLG [9].
We report a rare m.12334G>A variant in the mitochondrial (mt-) tRNALeu (CUN) (MT-TL2) gene with high levels of heteroplasmy in skeletal muscle. Our work proves that this represented a de novo mutational event and confirms the pathogenicity of this variant.
CASE REPORT
A 47 year old male with a history of migraine and glucose intolerance presented with bilateral moderate-severe ptosis which was first noticed incidentally in his late twenties. It progressed gradually and affected his activities of daily living; driving became more difficult, he also reported diplopia and difficulty reaching for objects. He suffered from painful cramps in his leg muscles at the end of the day.
He reported that four other family members had bilateral ptosis. The pedigree suggested possible autosomal dominant inheritance. On further review of family photographs, we confirmed apparent ptosis in three family members (Fig. 1).
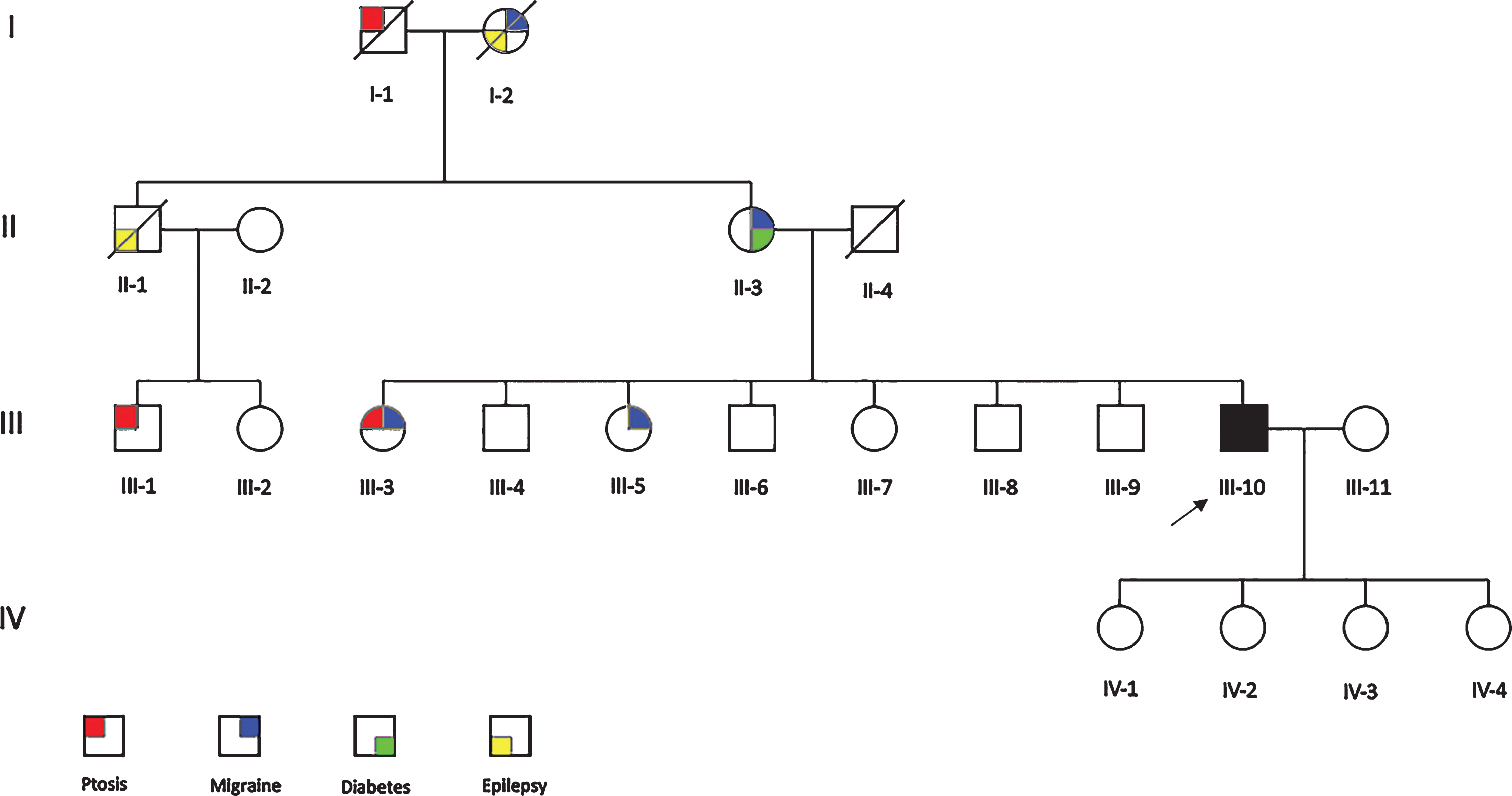
Family pedigree. The proband (III-10) is shaded in black.
Neurological examination revealed bilateral ptosis and ophthalmoplegia with reduction in all directions of gaze. The rest of the examination was normal. Blood investigations revealed a raised serum creatinine kinase of 499 U/L (normal <190 U/L). Other investigations were normal including venous blood lactate, electrocardiogram, 24 hour holter monitor, echocardiogram, brain magnetic resonance imaging and magnetic resonance angiography, nerve conduction studies (NCS) and electromyography (EMG).
We clinically assessed the patient’s mother (II-3) and two sisters (III-3 and III-5). III-3 reported ptosis that developed gradually in her early 50s. Examination revealed bilateral ptosis with normal extraocular eye movements and was otherwise unremarkable. All examined family members had normal NCS and EMG.
METHODS
Standard histological and histochemical analyses were performed on fresh-frozen sections (10 um) of skeletal muscle biopsy according to established protocols [10]. This included the assessment of cytochrome c oxidase (COX) both individually and using the sequential COX/succinate dehydrogenase (SDH) assay to identify respiratory-deficient fibres.
Total DNA was extracted from patient tissues and samples from other family members by standard procedures. The entire mitochondrial genome was amplified in two overlapping fragments by long-range PCR, and analysed by Next Generation Sequencing using an Ion Torrent™ Personal Genome Machine (PGM) platform (Thermo Fisher Scientific). Sequences were aligned to the revised Cambridge reference sequence (GenBank reference accession number: NC_012920.1) for human mtDNA [11]. Data analysis was performed in Torrent Suite v5.0.4 using Variant Caller v5.0.4.0 and Coverage Analysis v5.0.4.0.
Individual COX-positive and COX-deficient skeletal muscle fibres were isolated by laser microdissection and lysed to obtain total DNA to be used in single-fibre mutation segregation studies. The mtDNA mutation loads in homogenate tissues and individual COX-positive and COX-deficient fibres were determined by quantitative pyrosequencing, using mutation-specific primers (details available on request). Pyrosequencing was performed on the Pyromark Q24 platform according to manufacturer’s protocol. mtDNA heteroplasmy levels were quantified by comparing peak heights of wild-type and mutant nucleotides at the relevant position [12].
RESULTS
Given this gentleman’s clinical presentation, our initial diagnostic studies focussed on the targeted sequencing (using DNA derived from EDTA-blood) of 21 genes associated with disorders of mitochondrial DNA maintenance (ABAT, AFG3L2, AGK, TWNK, DGUOK, DNA2, FBXL4, MFN2, MGME1, MPV17, OPA1, POLG, POLG2, RNASEH1, RPM2B, SLC25A4, SPG7, SUCLA2, SUCLG1, TK2, TYMP) but this did not reveal causal or candidate pathogenic variants. We proceeded to a muscle biopsy which revealed type 2 fibre atrophy and numerous COX-deficient fibres in a mosaic pattern (Fig. 2A) with some evidence of fibres showing subsarcolemmal mitochondrial accumulation. Long range PCR analysis of a muscle DNA sample failed to reveal evidence of mtDNA rearrangements, prompting sequencing of the mitochondrial genome in muscle DNA, leading to the identification of a rare previously-reported m.12334G>A MT-TL2 variant [6]. The m.12334G>A variant was found to be present at high levels (84%) of heteroplasmy in the patient’s skeletal muscle DNA sample, with lower levels documented in urinary epithelium (24%), EDTA-blood (6%) and buccal epithelium (8%) using quantitative pyrosequencing.
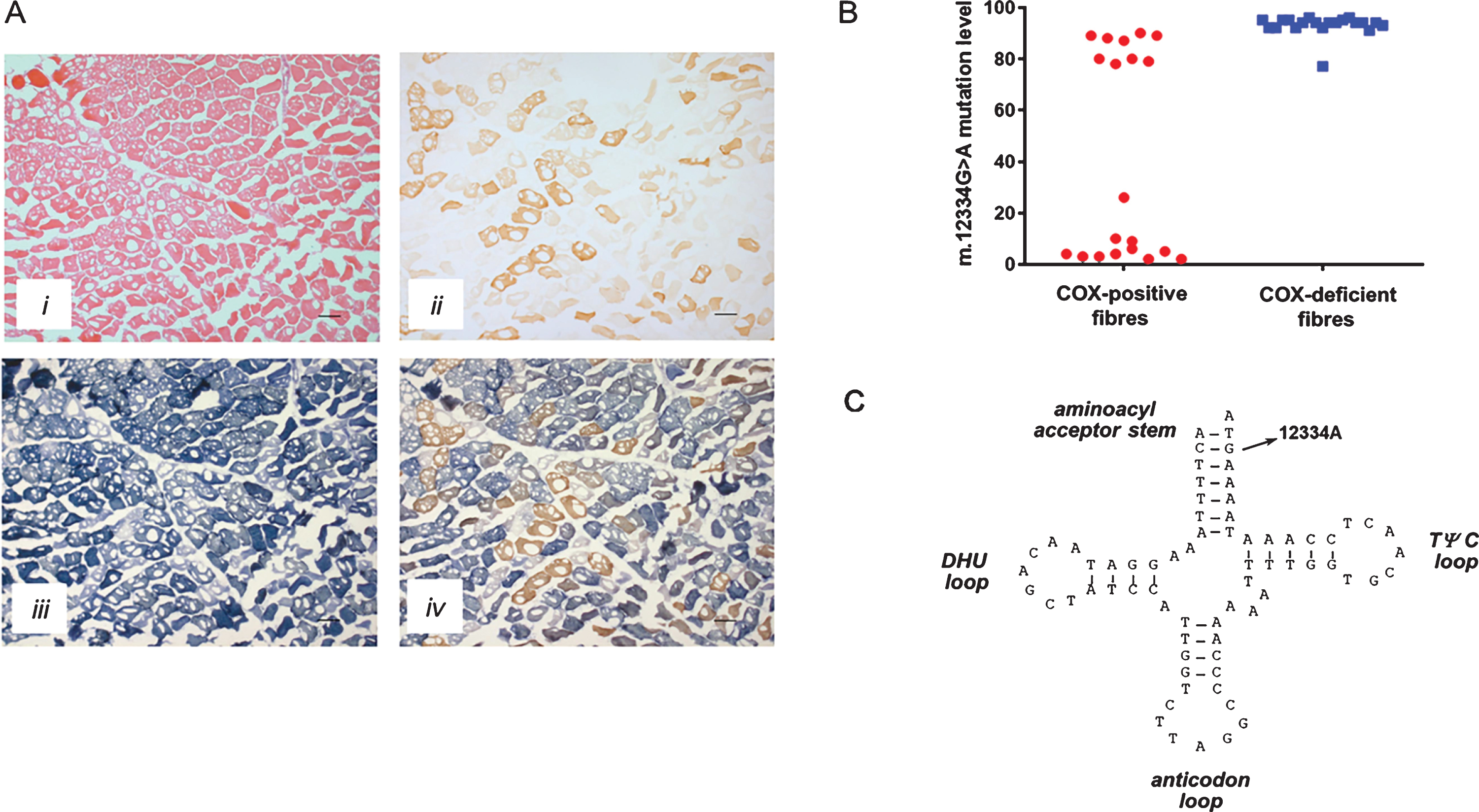
Histopathological and molecular evaluation of the patient. A, Histological and histochemical analyses of the patient’s skeletal muscle biopsy showing hematoxylin and eosin (H&E) staining (i), cytochrome c oxidase (COX) histochemistry (ii), succinate dehydrogenase (SDH) histochemistry (iii) and sequential COX-SDH histochemistry (iv), highlighting a marked mosaic pattern of COX deficiency whilst noting extensive freezing artefact throughout the biopsy; scale bar = 100μm.
Urinary, EDTA-blood and buccal epithelial DNA samples taken from the family members described above did not demonstrate any evidence of the MT-TL2 variant. Further review of family photographs and lack of consistent segregation between epilepsy, migraine and diabetes with the reported ptosis suggested that these features were less likely to be relevant in the family. In addition, patient III-3 had a muscle biopsy which did not demonstrate histocytochemical abnormalities suggestive of mitochondrial disease or any evidence of the m.12334G>A MT-TL2 variant.
In order to definitively confirm pathogenicity of the m.12334G>A MT-TL2 variant, single fibre quantitation of the mutation was performed in both COX-positive and COX-deficient muscle fibres to determine whether higher levels of mutated mtDNA co-segregated with a biochemical defect. Analysis of 20 COX-positive fibres showed a mean mutation load of 41.70±8.97%, whilst 20 COX-deficient fibres showed a mean mutation load of 92.95±0.90% (Fig. 2B, Unpaired T test p = <0.0001), thus confirming pathogenicity.
DISCUSSION
We describe a patient who presented with CPEO and muscle cramps with a reported family history initially suggestive of a possible autosomal dominant mode of inheritance. Although diabetes, epilepsy and migraine are over-represented in people with mitochondrial disease, they are also relatively common in the general population and, since they did not consistently segregate with ptosis in the pedigree, are likely not part of the inherited syndrome in this family. Prompted by the finding of extensive mitochondrial pathology in muscle, sequencing of the entire mitochondrial genome in muscle DNA revealed a rare m.12334G>A variant in the mitochondrial (mt-) tRNALeu (CUN) (MT-TL2) gene. Testing of family members proved this to be a de novo mutation. Furthermore, the patient’s sister (III-3), who had ptosis but no ophthalmoplegia, had a muscle biopsy which did not demonstrate any evidence of the m.12334G>A MT-TL2 variant. Pathogenicity was confirmed through single fibre analysis which demonstrated segregation of high m.12334G>A heteroplasmy levels with the COX histochemical defect.
The m.12334G>A MT-TL2 gene variant can be classed as pathogenic for several reasons. First, this is a rare variant and has only been reported once before in the literature [6]. We have not previously observed this variant in >1950 human mtDNA genomes in our in-house database of diagnostic human mtDNA sequences. Second, the variant is present at highest levels in clinically-affected tissue with a hierarchical segregation of the variant between different tissues; high heteroplasmy levels in skeletal muscle (84%) and low levels of heteroplasmy in urinary epithelium (24%), buccal epithelium (8%) and blood (6%). Third, this mutation appears to have arisen de novo based on mutation studies in family members, one of whom also had a muscle biopsy which did not demonstrate any evidence of the m.12334G>A MT-TL2 variant. Furthermore, the m.12334G>A variant is located within the amino acid acceptor stem of the mitochondrial tRNALeu (CUN) molecule, affecting an evolutionary-conserved nucleotide and is predicted to disrupt a Watson-Crick base pair within this stem which may alter tRNA secondary structure and impede function [6] (Fig. 2C). The most important evidence of pathogenicity is the single fibre quantitation study, which established that higher levels of mutated mtDNA co-segregated with a biochemical defect in the skeletal muscle tissue (Fig. 2B). Using the validated scoring system for assigning pathogenicity [13], this m.12334G>A variant also exceeds the required threshold to distinguish itself from polymorphic changes in the mt-tRNA, scoring 13 out of 20. The addition of this case to the previously reported one [6], together with our additional single fibre quantitation study strengthens the evidence in support of pathogenicity of this variant [13].
Mutations in MT-TL2 have also been reported to cause dilated cardiomyopathy, acquired idiopathic sideroblastic anaemia and mitochondrial myopathy [3, 4]. Similarly to the original case described by Vives-Bauza et al [6], our patient had normal NCS and EMG; however, unlike this case, we were able to perform a full neurological examination of some family members. Our patient also suffered from painful muscle cramps rather than exercise intolerance, had more severe bilateral ptosis with limitation of extraocular movements consistent with CPEO, had a raised creatinine kinase level and normal resting lactate. These differences demonstrate that the same variant can have variable clinical presentations. In addition, it highlights the genetic heterogeneity of CPEO which is more commonly seen in patients with single mitochondrial DNA deletions [8]. This adds to our growing understanding of the clinical and genetic heterogeneity in mitochondrial disease.
Interestingly, unlike the aforementioned case, our patient had low heteroplasmic levels of the mutation in other tissues (urinary epithelium, blood and buccal epithelium). In embryogenesis, buccal and urinary epithelium are derived from the endoderm whereas striated muscle and blood are derived from mesoderm [14]. Therefore we speculate that the mutation must have occurred at an early stage of embryogenesis. Somatic segregation of mitochondrial DNA which occurs randomly at various developmental stages may explain why we have found a differing tissue distribution of the mutation in our patient [15]. Moreover it is known that the mitochondrial mutation load can decline over time in rapidly dividing tissues such as blood due to selection against high levels of the mutated mtDNA [16].
This case confirms pathogenicity of the m.12334G>A variant and expands the phenotype of mutations in MT-TL2. Our case also highlights the importance of assessing family members when a patient reports a positive family history and of examining clinically-affected tissue in the proband.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to report.
Footnotes
ACKNOWLEDGMENTS
RWT is supported by the Wellcome Centre for Mitochondrial Research (203105/Z/16/Z), the Medical Research Council (MRC) International Centre for Genomic Medicine in Neuromuscular Disease, the MRC Mitochondrial Disease Patient Cohort: A Natural History Study and Patient Registry (G0800674), the Lily Foundation, the UK NIHR Biomedical Research Centre for Ageing, the Newcastle upon Tyne Foundation Hospitals NHS Trust, the MRC/EPSRC Molecular Pathology Node and the UK NHS Highly Specialised Service for Rare Mitochondrial Disorders of Adults and Children. We thank the Oxford University Hospitals Genetics Laboratory who performed the diagnostic gene panel testing for mtDNA maintenance disorders.