
Abstract
Background:
Insufficient amounts of survival motor neuron protein is leading to one of the most disabling neuromuscular diseases, spinal muscular atrophy (SMA). Before the current study, the detailed characteristics of Iranian patients with SMA had not been determined.
Objective:
To describe the key demographic, clinical, and genetic characteristics of patients with SMA registered in the Iranian Registry of SMA (IRSMA).
Methods:
IRSMA has been established since 2018, and the demographic, clinical, and genetic characteristics of patients with SMA were recorded according to the methods of treat neuromuscular disease (TREAT-NMD) project.
Results:
By October 1, 2022, 781 patients with 5q SMA were registered. Of them, 164 patients died, the majority of them had SMA type 1 and died during the first 20 months of life. The median survival of patients with type 1 SMA was 23 months. The consanguinity rate in 617 alive patients was 52.4%, while merely 24.8% of them had a positive family history. The most common type of SMA in live patients was type 3. Morbidities were defined as having scoliosis (44.1%), wheelchair dependency (36.8%), tube feeding (8.1%), and requiring mechanical ventilation (9.9%). Most of the registered patients had a homozygous deletion of SMN1, while the frequency of patients with higher copy numbers of SMN2, was less in more severe types of the disease. Earlier onset of the disease was significantly seen in patients with lower copy numbers of SMN2. The neuronal apoptosis inhibitory protein (NAIP) gene deletion was associated with a higher incidence of more severe types of SMA, higher dependency on ventilators, tube feeding, and earlier onset of the disease.
Conclusions:
The IRSMA is the first established Iranian nationwide registry of patients with SMA. Using this registry, decision-makers, researchers, and practitioners can precisely understand the epidemiology, characteristics, and genetics of patients with SMA in Iran.
INTRODUCTION
Spinal muscular atrophy (SMA), one of the most disabling autosomal recessive neuromuscular diseases (NMD), is characterized by degeneration of anterior horn cells of the spinal cord and results in progressive muscle atrophy and weakness. SMA is a rare condition, and one out of every 10,000 newborns can be affected by SMA [1, 2]. One in every fifty of the normal population (heterozygote frequency) carries the defective gene. Although the carrier frequency is different among different ethnicities, Caucasians have the highest [3]. The phenotypic spectrum of SMA is diverse, from severe hypotonia and generalized weakness at birth to extremely mild symptoms in adults [1]. Patients with SMA are affected by homozygous deletions (usually) or point mutations (infrequently) in exon 7 of the telomeric copy of the survival motor neuron gene (SMN1) at locus 5q13. In the absence of SMN1, the existing number of SMN2 copies determines disease severity [4, 5].
The absence or insufficient amounts of survival motor neuron (SMN) protein leads to spinal muscular atrophy in patients. Two genes (SMN1 and SMN2), which are highly similar, encode the full-length SMN protein. The SMA-determining gene is the SMN1 gene, which is absent or altered in patients with SMA. SMN2 gene, an extremely similar gene, produces low levels of SMN protein and allows survival in the absence or abnormal function of the SMN1 gene. Healthy people, carry at least one copy of telomeric SMN1 and one copy of centromeric SMN2 on chromosome 5 [5]. While the SMN1 gene is transcribed into a full-length SMN protein, the SMN2 gene produces merely 10% of the full-length SMN protein because of cytosine to thymine substitution in exon 7. This substitution causes exon 7 to be excluded from mature mRNA, resulting in a truncated protein (Δ7SMN) that is unstable and has quite a little function [1, 4].
The majority of patients who are affected by the most frequent type of SMA, type 1, die within the first two years of life. Patients with SMA type 2 present from 6 to 18 months of life and mainly develop progressive muscle weakness and contractures. Patients with SMA type 3 present after 3 years of age and usually have a normal life expectancy. According to the age of presentation, this type is divided into two subtypes; SMA type 3a, which presents before 3 years of age, and SMA type 3b, which presents afterward. Patients with the mildest type of SMA, type 4, merely show mild muscle weakness during adulthood. Patients with SMA type 4 retain the ability to walk and have a normal life expectancy [6].
To determine a trial- or treatment-ready population of patients with rare diseases such as SMA, registries are highly recommended. However, to describe long-term clinical courses for these kinds of diseases, we need natural course studies. The TREAT-NMD project (https://treat-nmd.org/), established in 2007, aimed to construct a global database of patients in the form of national registries as an important part of the TREAT-NMD project’s goals [7]. The cross-sectional outcomes and benefits of these worldwide partnerships have been established [8, 9].
According to the TREAT-NMD protocols, the Iranian Registry of SMA (IRSMA) was established in 2018 by the Tehran University of Medical Sciences (TUMS). After establishing IRSMA, the data of patients with SMA from all over the country have been collected, curated, and updated. In this report, we aimed to describe the key demographic, clinical, and genetic characteristics of patients included in the IRSMA.
MATERIALS AND METHODS
Iranian Registry of SMA (IRSMA)
The Iranian registry of SMA was aimed to determine the epidemiological, clinical, and genetic characteristics of Iranian patients with SMA. IRSMA also aimed to estimate the burden of SMA on the health care system, which is essential for planning patient support programs. Furthermore, identifying patients with SMA could help to provide the recently approved treatments to Iranian patients with SMA. Therefore, in 2018, IRSMA was established according to the goals of the international TREAT-NMD project. In addition to the patients that had been registered by the Iranian SMA association and Iran’s muscular dystrophy association, we registered the data of patients referred to more than twenty academic neuromuscular referral centers to cover all patients with SMA from all over the country. Since then, the patients’ data have been collected in a centralized registry, curated, and updated. The Iranian SMA association, a non-governmental organization (NGO) that was established years earlier, had the largest registry of patients with SMA in the country, given the supportive roles that this NGO has had for patients. The Iranian SMA registry is designed on a three-layer web-based data collection system and is based on the District Health Information Software 2 (DHIS2) - an open-source software platform for reporting, analysis, and dissemination of data - localized in the Children’s Medical Center, Pediatrics Center of Excellence, TUMS, Tehran, Iran (http://ismar.ir/dhis-web-commons/security/login.action). All the centers could register patients in the registry; however, registry coordinators supervise the accuracy and entirety of all records. All the data remained confidential in IRSMA and anonymous data were used for analysis by principal investigators.
Ethical issues
The research ethics committee of the TUMS (IR.TUMS.VCR.REC.1398.656) approved this study. The study was conducted according to the ethical standards laid down in the declaration of Helsinki of 1964 and its later amendments.
Data collection and entry method
The patients suspected of having SMA were referred for genetic tests. The patients with proven deletion of SMN1 – either homozygous or heterozygous with complex mutations –were entered into the registry. Patients’ data were collected through paper and electronic questionnaires. After obtaining informed consent, these questionnaires were completed by practicing physicians or trained personnel. The questionnaire included highly recommended items, as proposed by TREAT-NMD in 2007 and an extended version in 2018 [10, 11]. Data comprise information on demographic, clinical, genetic, and survival characteristics. The main variables were age, vital status, weight, date of birth, date of disease onset, date of diagnosis, date of death, type of SMA, disabilities - including requiring mechanical ventilation, tube feeding, and wheelchair dependency- comorbidities e.g., scoliosis, type of SMN1 deletion (i.e., homozygous, heterozygous with point mutations), the copy number of SMN2 exon 7 and exon 8, and neuronal apoptosis inhibitory protein (NAIP) gene deletion. The age of patients at disease onset, diagnosis, and death was calculated from the aforementioned dates. Genetic confirmation of SMA was a requirement for entrance into the registry, however, assessment of SMN2 copy number was highly recommended. SMN2 copy numbers were measured by multiplex ligation-dependent probe amplification (MLPA) assay. If necessary, patients or genetic laboratories were contacted for reevaluation and data clarification. Given that the genetic test for SMN2 copy numbers has been carried out in several laboratories in Iran just since a few years ago, a patient might have proven SMA without a measured SMN2 copy number. Whole exome sequencing (WES) was done for patients with suspicious clinical presentation without abnormality of the SMN1 gene (i.e., non-5q SMA patients). Moreover, the data for NAIP gene deletion (evaluated by polymerase chain reaction) were available in some of the laboratory reports. Patients were encouraged to update their data each year, and all the available data were updated before the publication of this report. Patients with incomplete data were finally excluded from statistical analysis. The accuracy of the data obtained using the questionnaires and compatibility with the diagnosis were verified by neurologists. Data were then entered manually into a central server computer at the TUMS with an option to transfer to Microsoft Excel® for analysis when needed.
Statistical analysis
Categorical variables were presented as sum and relative frequency, while continuous variables were described as mean and standard deviation or median and (5–95) percentiles. Comparisons between two groups of patients were carried out using an independent t-test, while comparisons between more than two groups were done using the ANOVA test. The comparison of categorical variables between the two groups was done using the chi-square test. A linear regression test was used to evaluate the correlation of different types of SMA with the frequency of clinical characteristics. Kaplan–Meier model was used to assess the survival of patients across different periods. A P-value of <0.05 was considered statistically significant. Data were analyzed using IBM SPSS® statistical software (version 24, SPSS Inc., Chicago, IL, USA) and graphs were illustrated using GraphPad Prism® (version 9.0.0 for Windows, GraphPad Software, San Diego, CA, USA), and Microsoft Excel® (2021, Redmond, WA).
RESULTS
Demographic information
In total, 789 patients were included in the registry on October 1, 2022. Eight patients (1%) diagnosed with non-5q SMA were excluded from the analysis, however, a brief clinical and genetic description of them was presented in supplementary Table 1. After excluding non-5q SMA patients, 781 patients with 5q SMA were included in the main analysis. Of them, 164 patients were dead and 617 of them were alive (Fig. 1). The mean age of the patients was 13.5±13.9 years, while the mean age of the alive and dead patients was 16.6±14.0 and 1.8±2.8, respectively. The male-to-female ratio was 1.2 (417 males and 364 females). There was a significant difference in age, gender, and weight between alive and dead patients (P < 0.001, P = 0.006, P = 0.004, respectively). Most of the patients who died had SMA type 1. Among the alive patients with SMA, 125 (20.3%), 163 (26.4%), 192 (31.1%), 94 (15.2%), and 43 (7%) had SMA type 1, 2, 3a, 3b, and 4, respectively (Table 1).
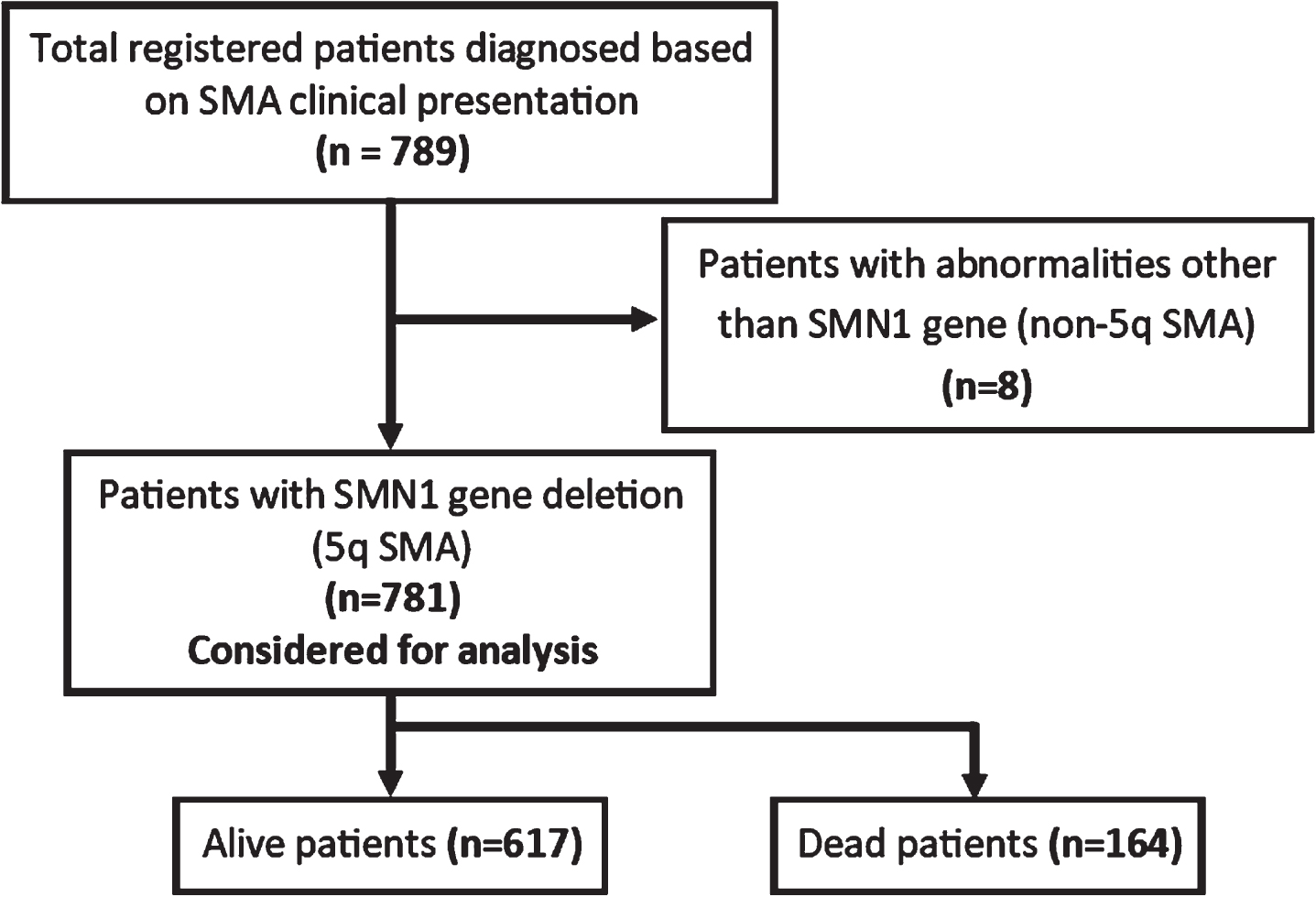
Flowchart of registered patients in the IRSMA. The patients with non-5q SMA were excluded from the main analysis.
Characteristics of patients included in the registry
*The quantitative data are shown as mean (standard deviation) and the qualitative data are shown as frequency (percentage).
The age of alive patients was calculated as the difference between the first of October 2022 and their birth date, while the age of dead patients was calculated as the difference between death and birth dates. Most deaths have occurred in the first two years of life. On the other hand, there was a decline in the relative frequency of alive SMA patients from the first age group to 20 years, after which there was a plateau to 45 years. The number of SMA patients older than 50 years old was extremely low, and no patient older than 70 years has been registered before this report (Figs. 2, 3).
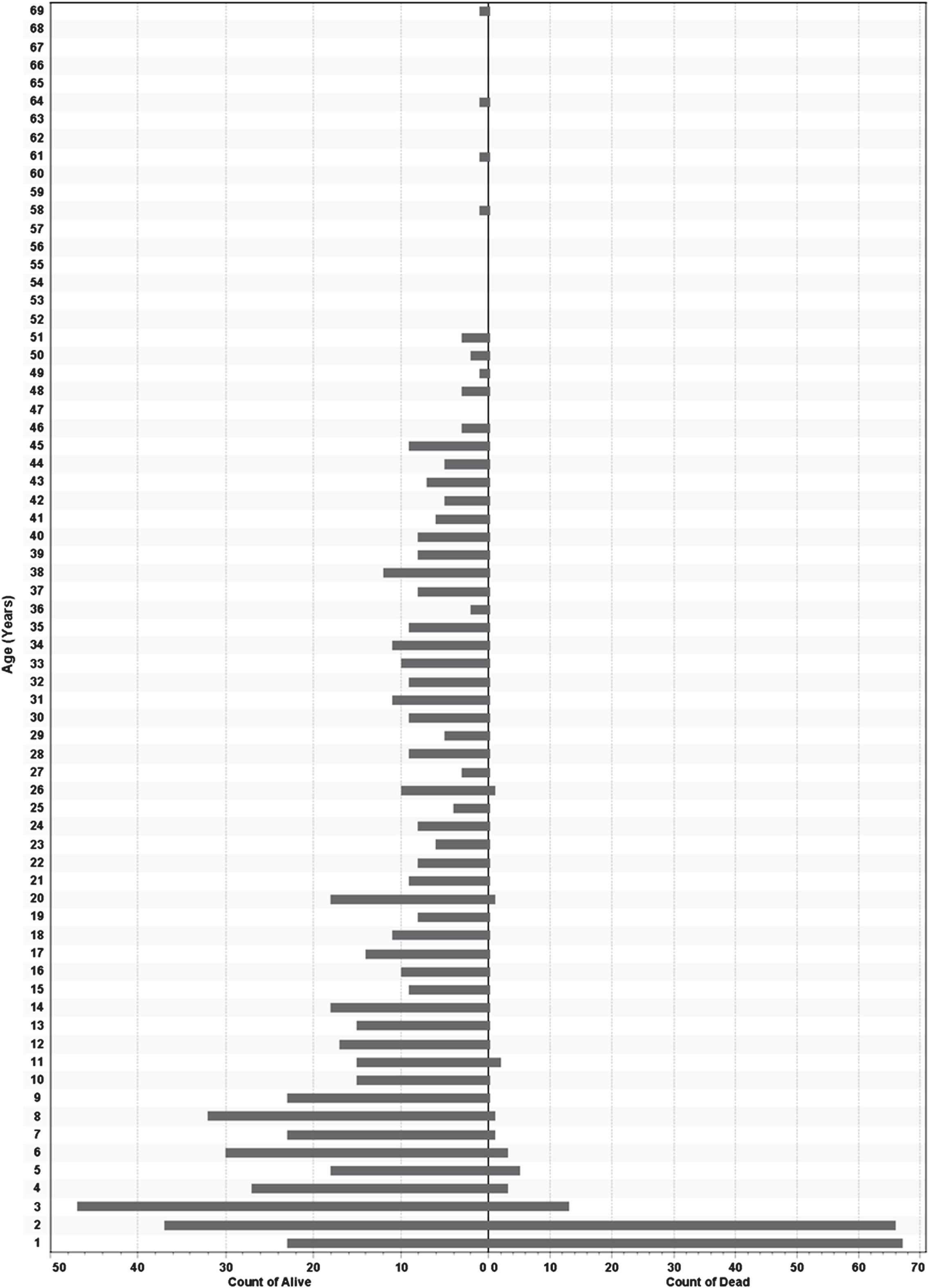
Population pyramid for alive (left) and dead (right) patients in the registry. Note that the frequency of each group is plotted.
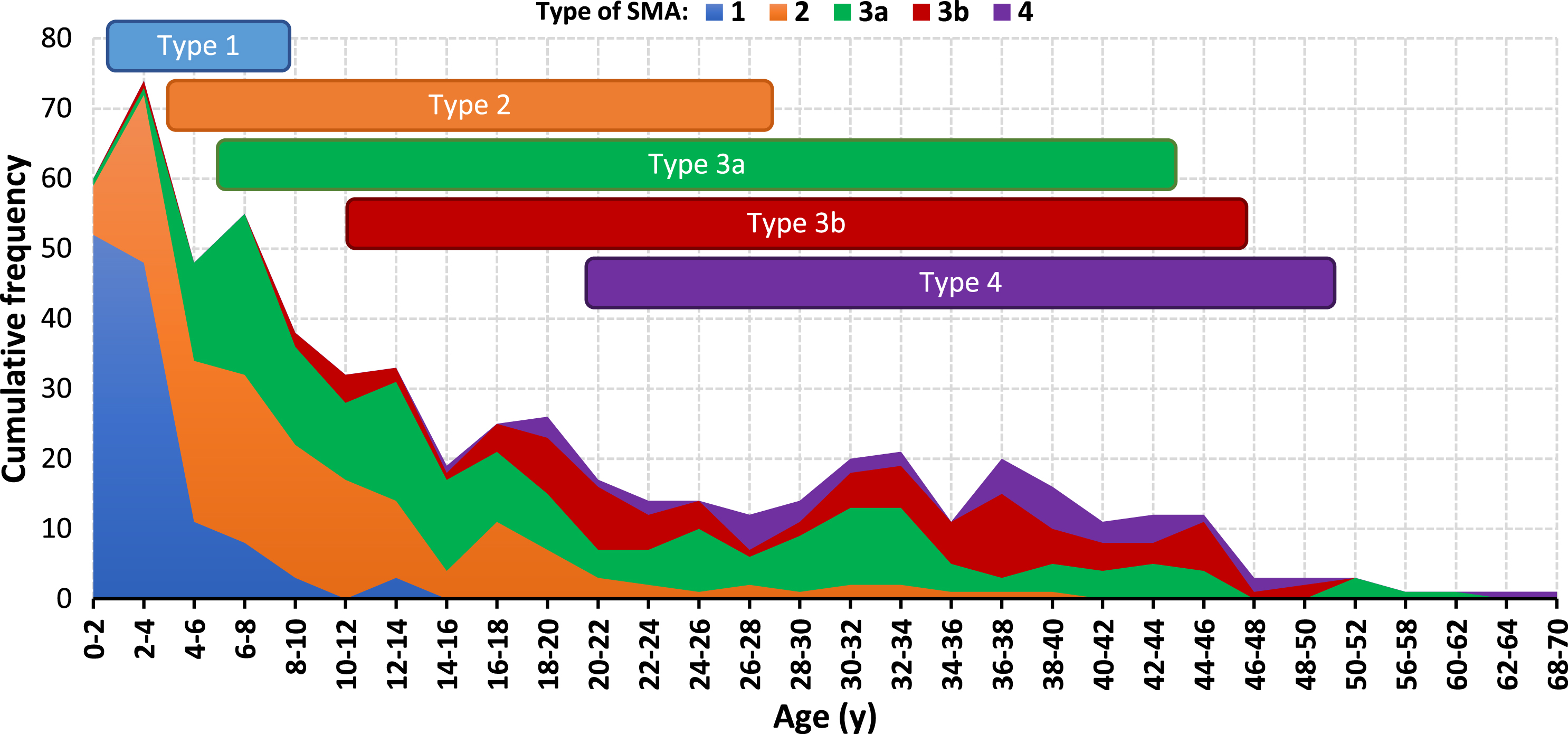
The distribution of SMA types across the different ages among the alive patients in the registry. The 5–95 percentiles range of age for each type of SMA are also shown.
The majority (5–95 percentiles) of type 1 SMA patients were under eight years, while the majority (5–95 percentiles) of the patients with type 2 SMA were between two and 27 years old. The age range of the majority (5–95 percentiles) of type 3a SMA patients started at five, while the age range for type 3b SMA and type 4 SMA patients started at approximately 10 and 20 years of age, respectively. These three latter SMA types (i.e., 3a, 3b, and 4) had almost similar highest achieved ages, given that the age range of the majority (5–95 percentiles) of patients with these types of SMA ended at 43, 45 and 49 years of age for type 3a and 3b and 4, respectively (Fig. 3).
Mortality and survival
Since the majority of deceased individuals had type 1 (98.8%), followed by type 2 (1.2%), we restricted the survival analysis to type 1 SMA. According to the Kaplan-Meier curve, the survival probabilities of type 1 SMA patients were 75.2%, 47.7%, 41.5%, 38.9%, and 33.3% at 1-year, 2-year, 3-year, 4-year, and 5-year, respectively. The median overall survival of patients with type 1 SMA was 23 months. More than 75% of dead patients have died before two years (Fig. 4).
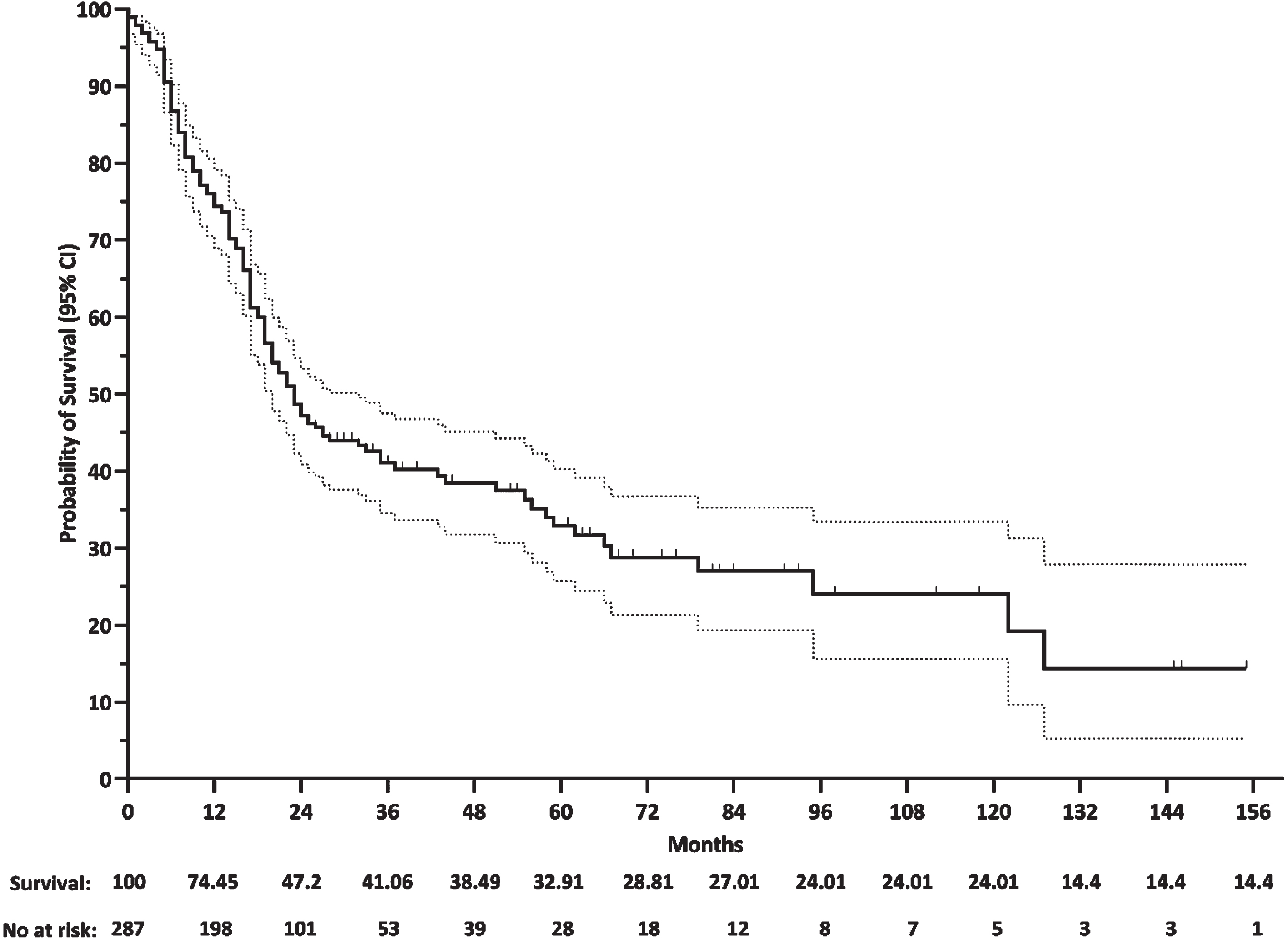
Kaplan-Meier curve for patients with SMA type 1.
Clinical characteristics
Assessing the clinical history of the alive patients and their disabilities, showed that most of the accessible patients were not dependent on mechanical ventilation for breathing (9.9%), tube for feeding (8.1%), and wheelchair for movement (36.8%). Furthermore, 44.1% of patients were affected by scoliosis. After drilling down the results to different SMA types, a significant trend in using mechanical ventilation and the feeding tube was observed, in which the more severe the type of SMA (moving from type 4 toward type 1), the higher the rate of dependency on mechanical ventilation and feeding tube (P < 0.001 and P < 0.001). Wheelchair dependency and scoliosis rates were relatively low in SMA types 1 and 4, while they were higher in patients with SMA types 2, 3a, and 3b (P < 0.001 and P < 0.001). Among the alive patients, the rates of consanguinity and positive family history were 52.4% and 24.8%, respectively. The rate of consanguinity and positive family history were significantly different among different types of SMA (P < 0.001 and P < 0.001). We also evaluated the patients respecting their age at first presentation, their age when they had the genetic test, and the interval between the onset of disease to diagnosis –also known as delay in diagnosis. In total, the mean age of onset was 44.9±68.6 months, while the mean age at diagnosis was 142.3±156.3 months. One of the patients was diagnosed prenatally, while four (0.7%) patients were diagnosed before disease onset. The average time interval from onset to diagnosis- delay in diagnosis- was 98.2±126.2 months. Drilling down the results to different types of SMA, there was a significant trend regarding the age of onset and age at diagnosis, in which patients with less severe types of SMA (moving from SMA type 1 to type 4), significantly had a later onset of disease and were diagnosed later (P < 0.001 and P < 0.001). On the other hand, the patients with less severe types of SMA significantly had a longer delay in diagnosis (B (95% CI) = 52.8 (46–58.7), P < 0.001) (Table 2).
Demographic and clinical characteristics of included alive patients by type of SMA
*The quantitative data are shown as mean (standard deviation) and the qualitative data are shown as frequency (percentage).
Genetic characteristics
All the patients had a deletion of SMN1. Among the alive patients (n = 617), 603 of them were affected by homozygous deletion. Amongst the rest (n = 14), one was affected by one homozygous point mutation in both alleles of SMN1, one was affected by two homozygous point mutations in both alleles of SMN1, and 12 were affected by compound heterozygous mutations with heterozygous deletion of one of the SMN1 alleles. Amongst patients with homozygous deletion (n = 603), 543 (90.1%) were affected by deletion of both exon 7 and 8 of SMN1, while 58 (9.6%) of them were affected by a homozygous deletion of exon 7 of SMN1 and two (0.3%) of them were affected by a homozygous deletion of exon 8 of SMN1. Notably, there are two patients with type 3a SMA with preserved exon 7 and homozygous deletion of exon 8 of SMN1 (Table 3).
Genetic characteristics of included alive patients by type of SMA
*The data are shown as frequency (percentage) **The data for copy number of exon 7 and exon 8 of SMN2 were available in 476 patients out of 617 alive patients. ***The data for NAIP gene were available in 131 patients out of 617 alive patients.
The data for copy numbers of exon 7 and exon 8 of SMN2 were available in 476 patients. The majority of SMA patients (76.5%) had two, or three copies of exon 7 of SMN2. Likewise, most of the patients (76.7%) had two, or three copies of exon 8 of SMN2. An equal number of copies of exons 7 and 8 of SMN2 was seen in 91.8% of patients. In 12 patients the copy numbers of exon 8 were higher than exon 7, while in 27 patients, it was vice versa (Table 3). Patients with milder types of SMA had higher copy numbers of exons 7 and 8 of the SMN2 gene (P < 0.001, P < 0.001, respectively). Similarly, the relative frequency of patients with one or two copy numbers was decreased from more severe types to milder types (P < 0.001, P < 0.001, respectively) (Fig. 5).
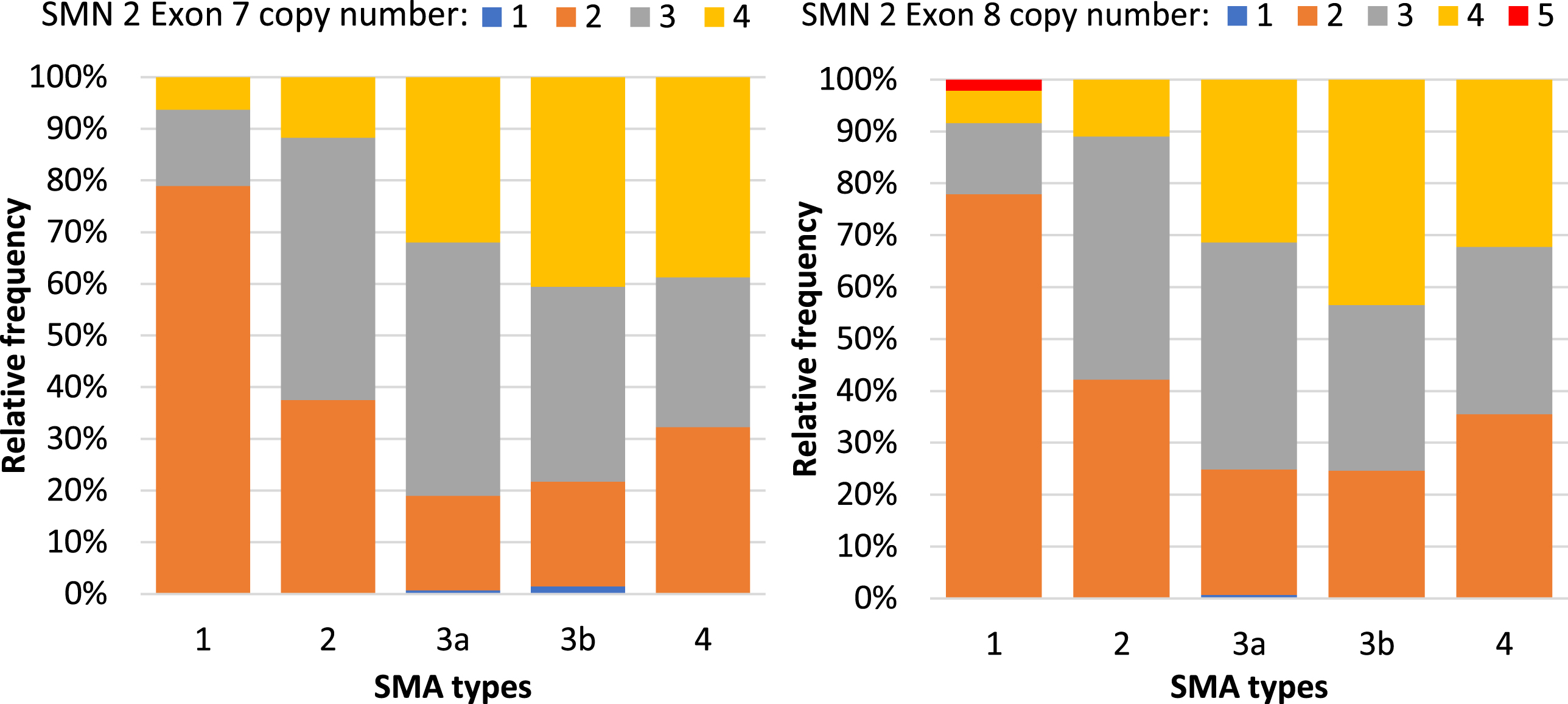
Distribution of the copy numbers of SMN2 exon 7 (left) and exon 8 (right) gene among the patients with available data included in the registry (n = 476).
Finally, we assessed the association between the copy numbers of exon 7 and exon 8 of SMN2 with age at the onset of the disease. Given the low frequency of patients with one or five copy numbers, they were excluded from the analysis. According to the results, patients with two copies of exon 7 of SMN2 presented at a significantly lower age compared to patients with four copies of exon 7 of SMN2 ((29.8±69.3 vs 70.6±74.4), P < 0.001). Similarly, patients with three copies of exon 7 of SMN2 had significantly more anticipated age of onset, compared to those with four copies ((41.9±59.9 vs 70.6±74.4), (P < 0.001)). The case was the same about the exon 8 copy numbers, in which patients with two copies of exon 8 of SMN2 had a significantly lower age at onset compared to those with four copies of exon 8 of SMN2 (32.8±70.4 vs 73.4±76.2, P < 0.001). Likewise, patients with three copies of exon 8 of SMN2 had a significantly earlier age at onset compared to those with four copies of exon 8 of SMN2 (38.9±55.6 vs 73.4±76.2, P < 0.001).
The distribution of NAIP gene deletion was significantly different across different types of SMA (P < 0.001) (Table 3). The frequency of NAIP deletion was higher in more severe cases compared to the milder types (e.g., type 1 vs type 4: 90.9% vs 0%, P < 0.001). Given that the NAIP deletion and SMN2 copy number were moderately correlated (B = –0.411, P < 0.001), we categorized the patients into groups based on their status of NAIP deletion and copy numbers of exon 7 of SMN2 (Table 4). Considering NAIP and SMN2 genotype combinations, the distribution of SMA types was significantly different across different genotype combinations (P < 0.001) (Table 3) (Supplementary Figure 1).
Clinical characteristics of alive patients across NAIP-SMN2 genotype combinations
*The quantitative data are shown as mean (standard deviation) and the qualitative data are shown as frequency (percentage). **Calculated by chi-square test comparing patients with/without NAIP deletion. ***Calculated by chi-square test comparing patients with different NAIP-SMN2 genotype combinations. *****Calculated by Mann-Whitney U test comparing patients with/without NAIP deletion. ******Calculated by Kruskal Wallis test comparing patients with/without NAIP deletion.
A significant correlation was seen between the deletion of the NAIP gene and clinical features of alive SMA patients who had available data for the NAIP gene. Patients with NAIP deletion had a higher rate of dependency on ventilator and tube feeding along with the earlier onset of the disease (P = 0.006, 0.005, and <0.001, respectively), meanwhile the rate of wheelchair dependency was lower in patients with NAIP deletion (P = 0.045) (Table 4). Considering NAIP and SMN2 genotype combinations, the results were nearly the same i.e., the rate of scoliosis and dependency on ventilator, wheelchair, and tube feeding were significantly different across different genotype combinations (P = 0.007, <0.001, <0.001 and 0.044, respectively). The average age at onset was also significantly different across the combinations (P < 0.001) (Table 4).
DISCUSSION
To the best of our knowledge, just three registries from Turkey [12], Lebanon [13], and Egypt [14] among all the Middle East countries have been planned to collect various data from patients with SMA. This is the first time that an Iranian registry, IRSMA, collects critical information regarding the demographic, clinical, and genetic characteristics of Iranian patients with SMA. The dataset was established based on the TREAT-NMD project, and recently a revision was planned in such a way that the registry could be compatible with updates in standards of care and map emerging treatments. Currently, data from 781 patients with different types of 5q SMA (mean age of 13.5 years, range of age of 0 –68.3 years) has been collected in IRSMA. It is worth noting that the frequency of non-5q SMA patients (n = 8) was one percent in IRSMA and the most common genetic underlying cause among them was ASAH1. These data are a bit different from other studies, reporting a frequency of up to four to five percent of non-5q SMA patients. It is mainly due to the low accessibility and the high price of whole exome (WES) and whole genome (WGS) sequencing which hinder the further evaluation of patients with normal MLPA tests.
The majority of registered patients in IRSMA were under 20 years (75th percentile = 21.3 years). This finding was consistent with the age range of SMA patients in several other registries [8]. Bladen et al. conducted a survey of 24 national registries in North America, Europe, and Australia including a total of approximately 5,000 patients with SMA. The number of patients from birth to 20 years of age increased and after that, the number of patients in the registries steadily decreased [8]. However, in our registry, there was a decline in the number of SMA patients from birth to 20, after which there was a plateau to 45 years. Likewise other registries, the number of SMA patients older than 50 years was extremely low [8, 15]. This higher percentage of under-20-year-old patients may be because of the recent emergence of genetic diagnosis procedures, the increasing number of laboratories providing these services even in underprivileged areas, and most importantly, increased public awareness in Iran.
According to the reports of several other registries, the subtype with the smallest prevalence was SMA type 1. However, it had the highest incidence among the SMA subtypes [6, 15]. Likewise, in IRSMA, we recorded a large number of SMA type 1 patients (about 37% across the whole and 20% in alive patients). Although it is noteworthy that more than half of them were deceased at the time of this report, we succeeded to register the SMA type 1 cases as much as possible in our registry, given the widespread and strong referral system in Iran. However, there is a long way to become close to the actual incidence of type 1 SMA (i.e., approximately 60%) [16]. Higher severity and shorter survival of type 1 SMA were reflected in the age distribution of patients to some extent, which could justify the decrease in the number of SMA patients from birth to 20 years in our records. Among all alive patients included in IRSMA, almost 45% of patients had SMA type 3. Several countries including Poland, France, Hungary, Romania, Bulgaria, USA, and UK also registered the largest patient numbers of type 3 SMA in their registries, while SMA type 2 was the main type with the largest number of patients in Canada, Germany, Spain, Switzerland, Italy, Ukraine, Austria, Argentina, Mexico, New Zealand, Czech Republic, and Turkey registries [8, 17].
At the time of this report, almost 80% of the registered patients were alive. The majority of deceased individuals (98.8%) were affected by type 1, followed by type 2 (1.2%) in our registry. The distribution of deceased individuals was similar to the “Cure SMA” database, which already has been presented by Belter et al. [18].
In our registry, based on the survival analysis the survival probabilities of type 1 SMA patients were 75.2%, 47.7%, and 38.9% at one-year, two-year, and four-year of age, respectively. In a recent population–based study among 32 patients with type 1c, the probability of survival was 97.7%, 94.9%, and 88.5% at one, two, and four years of age. Moreover, evaluating 25 patients with SMA type 1b showed a probability of survival of 30.6% at age of one year, decreasing to 6.5%, and 0.2% at two and four years of age [19]. Given the definition of types 1b and 1c, we should consider the cumulative results of these two types. In this way, this result was rather consistent with our findings. Although we did not categorize type 1 into its subtypes, it should be noted that the number of type 1 patients who were included in the analysis in our study was much larger than the previous studies [19–22].
The median survival of individuals with type 1 in IRSMA was 23 months, while other registries reported median survival ranging from 6 to 12 months [18, 21–29]. The factors contributing to the higher survival of Iranian SMA patients compared to other countries should be sought in the future studies, however, not including more severe cases, the existence of supporting communities and positive actions of social welfare systems could potentially explain this higher survival.
Survival of SMA type 1 who were born before 2018 (establishment of IRSMA) was significantly lower than those who were born after 2018 (e.g., 4-year overall survival of 29% vs. 59.9%, log-rank P < 0.001). A cohort study published in 2007 found that the survival of patients who were born in 1995–2006 was significantly higher than those who were born in 1980–1994 (median survival of 24 months vs. 7.5 months with a 70% reduction in the risk of death) [30]. This increase in survival over time shows that supportive care including the use of noninvasive respiratory support and gastrostomy tube feeding would provide adequate nutrition while preventing aspiration, resulting in a better quality of care. It strongly supports the necessity for early intervention by an experienced team for SMA patient care.
In our study, the rate of consanguinity was 52.4%, which was higher than other published reports of SMA patients [16]. The higher rate of consanguinity in Iran refers to the cultural and social characteristics of the Iranian population [31]. Several previous reports have also reported the rate of consanguinity in the Iranian population to be as high as 38.6%, while 27.9% of them were first-cousin marriages [32]. This higher rate of consanguinity among SMA patients was previously reported in middle east countries [14, 33–35]. The higher-than-average rate of consanguineous marriage could potentially increase the incidence and prevalence of SMA patients in these populations [16].
Like a previous study, we found a significant increasing trend in using mechanical ventilation and feeding tube from milder types to more severe types. On the other hand, the wheelchair dependency and scoliosis rates were relatively low in SMA types 1 and 4, while they were higher for patients with SMA types 2 and 3 [8].
An essential metric of SMA diagnosis is the delay in diagnosis. Corroborated with other registries, the average age of onset in our study was 3.5, 13.4, 31.4, 104.6, 214.9, and 44.9 months for types 1, 2, 3a, 3b, and 4, respectively, while the IRSMA database indicates that the mean age at diagnosis was 9.5, 64.9, 182, 284.6, 346.2 and 142.3 months for types 1, 2, 3a, 3b, and 4, respectively. The age of diagnosis of SMA in all types was extremely later than that reported in the “Cure SMA” registry in the US [18]. In addition, a recent systematic literature review showed similar results with “Cure SMA” [36]. The average age at onset and age at diagnosis across the time showed a dramatic decrease in delay in diagnosis in all types of SMA (Supplementary Figure 2). The average delay in diagnosis among the patients who have been born since 2018 - the year of establishment of the IRSMA- was prominently lower (108.2±129.5 vs 3.2±5.7, P < 0.001). This finding showed the improved early diagnosis of SMA in recent years in Iran and the efficiency of the healthcare system in identifying new cases as early as possible. The average age at diagnosis of over nine months of age for type 1 SMA, is particularly problematic since there is substantial evidence that in SMA type 1 the irreversible loss of motor neurons begins in the perinatal period, and is followed by significant degeneration in the first 3 months of life and loss of more than 90% of motor units by 6 months of age [37, 38]. This considerable delay in diagnosis, together with the higher efficacy of the early intervention [39], shows the need for, and the benefit of comprehensive unbiased screening of newborns for early diagnosis of SMA. Early treatment with novel therapies, which could only be possible with an early diagnosis, can enhance clinical outcomes in SMA patients [40]. Although Iran has a far way to include SMA in prenatal screening programs, growing awareness about this disease was remarkable during the history of IRSMA. Moreover, recently, the importance of pre–implantation and prenatal genetic screening tests in families with a history of SMA has been noticed more.
Among the patients who were registered in IRSMA, 97.7% of them were affected by homozygous deletion. Among patients with homozygous deletion, 90.1% of them were affected by a deletion of both exons 7 and 8 of SMN1. In general, these results were consistent with the results reported by Megarbane et al. from Lebanon that revealed the homozygous deletions of both exons 7 and 8 of SMN1 were the most common molecular finding among 90.7% of SMA patients (n = 185) [13]. Assessment of SMN2 gene copy number, which generally correlates inversely with disease severity [41], was limited to 77.2% of patients as it was not a standard protocol at the time of the IRSMA establishment. According to the data of alive patients, the most common copy numbers of exon 7 and exon 8 of SMN2 in our registry were two or three copies. Corroborated with previous studies, patients with milder types of SMA (i.e., moving from type 1 to type 4 SMA) generally had higher copy numbers of exons 7 and 8 of the SMN2 gene [18]. We also found that the patients with higher copy numbers of the SMN2 gene had a later disease age of onset.
It is worth noticing some patients with rather discrepant SMN2 copy numbers (e.g., patients with type 4 SMA with two copies or patients with type 3 SMA with just one copy or on the other hand type 1 SMA patients with 4 or 5 copies of SMN2). It can be hypothesized that other gene modifiers, which are not considered in routine MLPA assay, may also affect the severity of the disease [42–45]. We evaluated the effect of NAIP gene deletion, in which NAIP deletion had a deleterious effect i.e., higher incidence of more severe types of SMA, higher dependency on ventilator, tube feeding, and earlier onset of the disease. Considering the NAIP-SMN2 genotype combinations also resulted in the rather same outcome; of note, it seems that it can predict the SMA types better than SMN2 copy numbers alone (except for two patients in the NAIP del + 1 SMN2 copy number group) (Fig. 5 vs Supplementary Figure 1). In general, these findings are similar to the results of previous studies. Ahn et al found that NAIP deletion in SMA patients was associated with earlier onset, and worse outcomes (i.e., earlier mortality and need for mechanical ventilation support) in SMA patients [46]. Recent studies indicated that NAIP deletion was closely associated with the severity of SMA [47, 48]. In another study, SMN2 and NAIP were introduced as the most important modifier genes which copy numbers can affect the severity of SMA. They concluded that the combination of modifier genes is preferred to using just the copy numbers of exon 7 of the SMN2 gene to determine the prognostic pattern of SMA [49]. Additional population-wide investigations should be done for figuring out how the complex interplay of genetic modifiers influences SMA severity.
As with any rare disease registry, there is a recruitment bias; patients with SMA who reach out to IRSMA might not represent the whole SMA population. Being included in IRSMA might be limited to a more engaged population, particularly in families with a history of SMA. Those SMA patients who have not been diagnosed or those who live in remote areas with limited diagnostic facilities might be missed. Given the average annual number of births (18 per 1000 population) from 2018 to 2021 in Iran [9, 50], the average total population of Iran in this period (N = 86,848,847) [7, 51] and a worldwide SMA incidence of near 1 per 10,000 live births [16], we expected 635 SMA patients to be born in the registration period up to 2021. Respecting that there is no established perinatal screening program for SMA in Iran, we merely could have the chance to detect the patients who have become symptomatic in this period or the patients who voluntarily have taken a test for it (e.g., due to similar cases in their family). With an acceptable estimation (considering an incidence of 6 per 100,000 live births for type 1 SMA [16]), 381 type 1 patients should have been diagnosed in this period; while we have registered 175 type 1 SMA patients with birth dates within this period (approximately 45.4% of the expected patients). Self-reported data is susceptible to errors in memory and reporting inaccurate or incomplete information. SMN2 gene copy number was only available for 77.2% of the patients. The reason we missed some genetic information was the lack of a standard protocol for SMA genetic testing at the time of IRSMA establishment. Therefore, we advised all laboratories to report the results of new SMA cases according to the developed standard protocol to include the additional helpful genetic data in their report for the treatment. The included patients in IRSMA likely represent a broader portion of the population than a survey limited to a single or tertiary care center. As the IRSMA program covers all ages and types of SMA, we could include a more diverse group of patients compared to the programs conducted by children’s hospitals. The most prominent strength of IRSMA is that this was the first Iranian registry of SMA patients with a comparatively large number of patients as with other national registries [18]. Our next mission will be to widely register all people with SMA in Iran then we can provide them with new treatments and supportive care facilities. In addition, we aim to help policymakers plan for the unmet needs of SMA patients in Iran.
In conclusion, the IRSMA, the first nationwide registry of SMA patients, aimed to contribute to a better understanding of the epidemiology of SMA in Iran. We described the demographic, clinical, and genetic characteristics of Iranian SMA patients for the first time. It also has a critical role in providing access to clinical trials for patients. With the recent approval of treatments and many efforts underway for the development of other novel medications, these kinds of registries could help allocate and prioritize the treatments to appropriate candidates. In addition, using the results from this registry, patient support organizations could implement several activities to empower patients and their families, campaign for patients’ social rights, and lobby to provide more insurance and treatment coverage by the government.
Footnotes
ACKNOWLEDGMENTS
The authors would like to thank the IRSMA coordinators for maintaining the registry at its highest quality. The authors would also like to thank all the families and individuals who have been in IRSMA for providing invaluable data. This endeavor would not have been possible without the kind and constructive contributions of the TUMS registry secretariat, the Iranian SMA association, Iran’s muscular dystrophy association, Dr. Azin Nahvijou, Dr. Shahram Savad, Bita Hemati, Ali Sharifi Kia, Saeid Azamian, Ebrahim Rahimian, Ramak Heidari, Farnaz Pourkave, Leila Majidi Rozbahani.
FUNDING
This survey was funded by the research deputy of the Tehran University of Medical Sciences. (Project number: 43353-224-02-98)
CONFLICT OF INTEREST
Mahmoud Reza Ashrafi is the IRSMA founder and board member of the Iranian SMA association. Gholamreza Zamani is a board member of Iran’s muscular dystrophy association. Morteza Heidari is the curator of IRSMA. None of the other authors have conflict of interest to report.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.