
Abstract
Background
Spinal Muscular Atrophy (SMA) leads to motor neuron loss, with progressive muscle weakness and wasting. Nationwide registries for neuromuscular diseases are pivotal for assessing epidemiology, preparing for clinical trials, and for adopting standardized management guidelines.
Objectives
This paper aims to present data gathered during the establishment of Pakistan's inaugural registry for genetically confirmed SMA cases.
Methods
In this retrospective study, 215 participants with genetically confirmed SMA were recruited. Telephonic interviews were conducted to collect data for the Muscular Disease Registry of Pakistan that was analyzed using STATA version 17.0.
Results
SMA type 1 was the most common type (71.2%, n = 153). Amongst patients who were tested for survival motor neuron (SMN2) copies, the majority (84.4%, n = 168) had two SMN2 copies. SMA types were significantly associated with the ability to sit (p < 0.001) and walk (p < 0.001), and usage of a wheelchair (p = 0.0054). SMN2 copy numbers were significantly associated with the ability to sit (p = 0.020) and walk (p = 0.031).
Conclusions
This study highlights the high prevalence of SMA genotypes and phenotypes associated with severe disease in our population. Our findings reiterate the challenging prognosis for Pakistani children with SMA and underscore the necessity of the development of nationwide newborn screening programs and making treatments available.
Introduction
Spinal Muscular Atrophy (SMA) is an autosomal recessive disease characterized by progressive weakness and atrophy of muscles 1 caused by degeneration of lower motor neurons in the spinal cord and brainstem secondary to gene deletions and mutations of the survival motor neuron 1 (SMN1) gene. 2 Severity of SMA is influenced by the number of copies of the homolog gene SMN2 that an affected individual carries. The latter produces limited quantities of functional SMN protein, thus affecting the severity of the SMA phenotype.3,4 SMA is categorized into five types (types 0-4) based on the age of onset and motor function, with types 0, 1, and 2 being more severe. 5
The incidence of SMA is estimated to be 1 in 15,000 live births. 6 The prevalence and incidence of the disease vary by geographic location.7–15 A high prevalence of SMA has been reported in Middle Eastern and South Asian countries such as Morocco, Egypt, Saudi Arabia, Iran and Pakistan, hypothesized to be secondary to the higher rates of consanguinity in these countries,16–20 seen as high as 65% in Pakistan. 21
Despite being the sixth most populous country globally, Pakistan has no disease registry available to collect data on patients with neuromuscular diseases (NMDs). This is due to a number of factors, including a lack of a nationwide newborn screening program, scarcity of diagnostic resources and government funding on healthcare,22,23 limited genomic literacy in both the general population and healthcare professionals, shortage of a professionally-trained genomics workforce, and non-availability of epidemiological data. 24
This pilot study aimed to gather information on genetically proven SMA cases in Pakistan to create the country's first disease registry for Spinal Muscular Atrophy and Duchenne Muscular Dystrophy. Here, we report data regarding SMA patients. The registry will be crucial to understanding the epidemiology of SMA in Pakistan, facilitating in preparation for clinical trials for new treatments, advocating for treatment availability and utilization, development of newborn screening and adopting standardized care guidelines.
Methods
Setting and design
This retrospective observational study includes patients who underwent genetic testing for SMA between January 2007 to August 2023. Data collection occurred from April 2019 to May 2024.
In 2017, we began developing the Muscular Dystrophy Registry of Pakistan (MDR-PK, www.mdrpakistan.com). Funding for this project was received by the Neurology Awareness & Research Foundation (NARF) Pakistan. After receiving approval from the National Bioethics Committee of Pakistan, the Aga Khan University Ethics Review Committee (5369-Med-ERC-18), and the University of North Carolina at Charlotte Institutional Review Board (IRB 18-0444), data collection commenced in April 2019. The MDR-PK registry was incorporated into the TREAT-NMD platform in October 2023.
Study population
The clinical laboratory at our tertiary care center, the Aga Khan University Hospital (AKUH), is the only clinical laboratory in Pakistan with the capability to conduct genetic testing for SMA. Therefore, all individuals in Pakistan displaying symptoms suggestive of SMA are referred by their healthcare provider to our laboratory for genetic testing and confirmation. Some patients in this study were also referred for SMA genetic testing after a first-degree relative had tested positive for SMA. Patients were categorized into SMA types using the criteria previously described by Cartwright and Upadhya. The classification criteria takes into consideration the number of SMN2 copies, onset of SMA symptoms, and development of ability to sit and stand. 3
Participants of all ages, from birth onwards, who were diagnosed with SMA based on positive genetic testing results were included in this study. Those with negative genetic test results and fetuses diagnosed with prenatal SMA were excluded.
Data collection instrument and procedures
We modeled the data collection instrument on the TREAT-NMD questionnaire for spinal muscular atrophy. This instrument included domains related to personal history, results of prior investigations (such as electromyography and muscle biopsy), and results of genetic testing for SMA (including type of mutations in SMN1 gene and number of SMN2 copies). The questionnaire also included a section regarding previous medical history, such as SMA symptom onset, motor functionality, ambulation status, nutrition, and pulmonary function.
For patients whose genetic tests were conducted during the data collection period (April 2019 - May 2024), informed consent was taken at the time of testing at the molecular laboratory. Consenting parents/legal guardians of patients referred for SMA testing were informed that if their patient tested positive for SMA, a research assistant (RA) part of the study team would contact them through telephone call and administer the registry questionnaire. The parents/legal guardians of SMA patients whose genetic tests were conducted prior to the data collection period were contacted through phone call by the RA, who proceeded to collect data only after obtaining informed verbal consent from the patients’ parents/legal guardians.
Data collection commenced in 2019 and was conducted by a RA. The RA was responsible for contacting families of all patients with positive genetic test results through phone calls, administering the questionnaire, and recording responses. Responses were then uploaded to the Muscular Dystrophy Registry of Pakistan (MDR-PK) and, subsequently, to the TREAT-NMD platform in October 2023. Study procedures are illustrated in Figure 1.
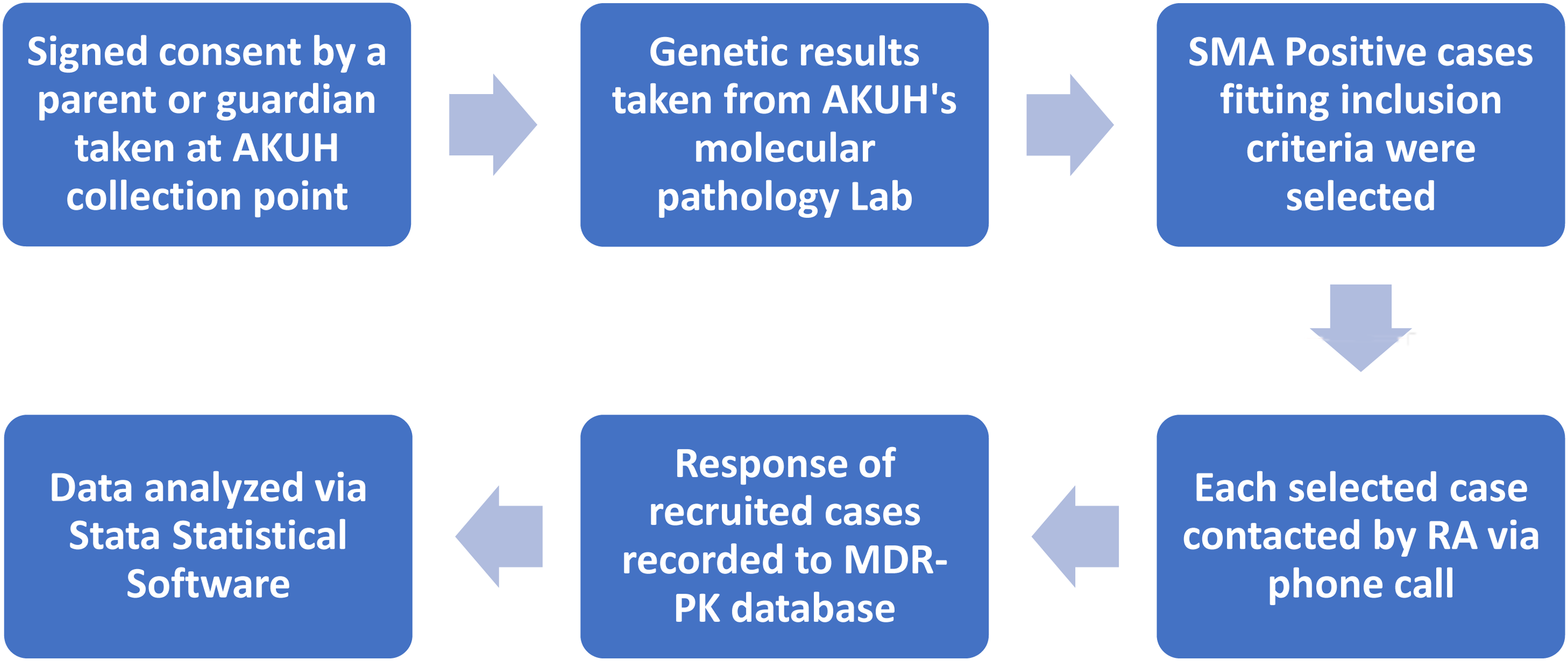
Study procedures - demonstrating steps taken to gather and process the data.
Genetic testing procedures
All SMA genetic testing was conducted by the Section of Molecular Pathology, AKUH Clinical Laboratories, Stadium Road, Karachi. The multiple probe-based ligation amplification (MLPA) method was used for the detection of SMN1 gene mutations to detect deletions in exons 7 and 8 of the gene and to evaluate the relative copy number of each DNA target. This also included identification of the SMN2 gene. The SALSA MLPA P021-A2 SMA, MRC Holland, Amsterdam, Netherlands kit was used. Notably, MLPA does not detect point mutations, which account for SMA in a minority of cases.
Data analysis
We summarized the demographic and genetic characteristics, such as number of SMN2 copies and SMA types using frequency distributions. Furthermore, we analyzed the association between various medical characteristics (such as the ability to sit and walk independently) and SMA types and SMN2 copy numbers using chi-square testing. Only patients with known SMA type and known SMN2 copy numbers were included in their respective analyses. In addition, to determine the association between consanguineous parentage with various patient variables, we used chi-square testing and Fischer's exact test where appropriate. A p-value below 0.05 was considered statistically significant. We analyzed data using Stata statistical software, version 17.0.
Results
We enrolled 215 SMA participants in this registry. The majority of our participants (62.8%, n = 135) had passed away by the time data was collected through telephonic interviews with parents/legal guardians. A large proportion of our participants were less than 1 years old (75.3%, n = 162) at the time of diagnosis. The majority of our patients (59.5%, n = 128) were children of consanguineous parents who were first cousins. SMA type 1 was the most common type in our population (71.2%, n = 153), followed by SMA type 2 (13.0%, n = 28). The median age at diagnosis was 4 months (2–11 months). The median age at diagnosis was lowest in SMA type 1 patients at 3.0 (2.0–50.0) months, and highest in SMA type 4 patients at 270.0 (147.0–335.3) months. The median age of participants who were alive at the time of interview was 27 months (7–144 months) (Table 1). None of our patients received disease-modifying treatment for SMA. The median age at death was calculated to be 6 months (4–9 months).
Demographic characteristics of SMA patients (n = 215).
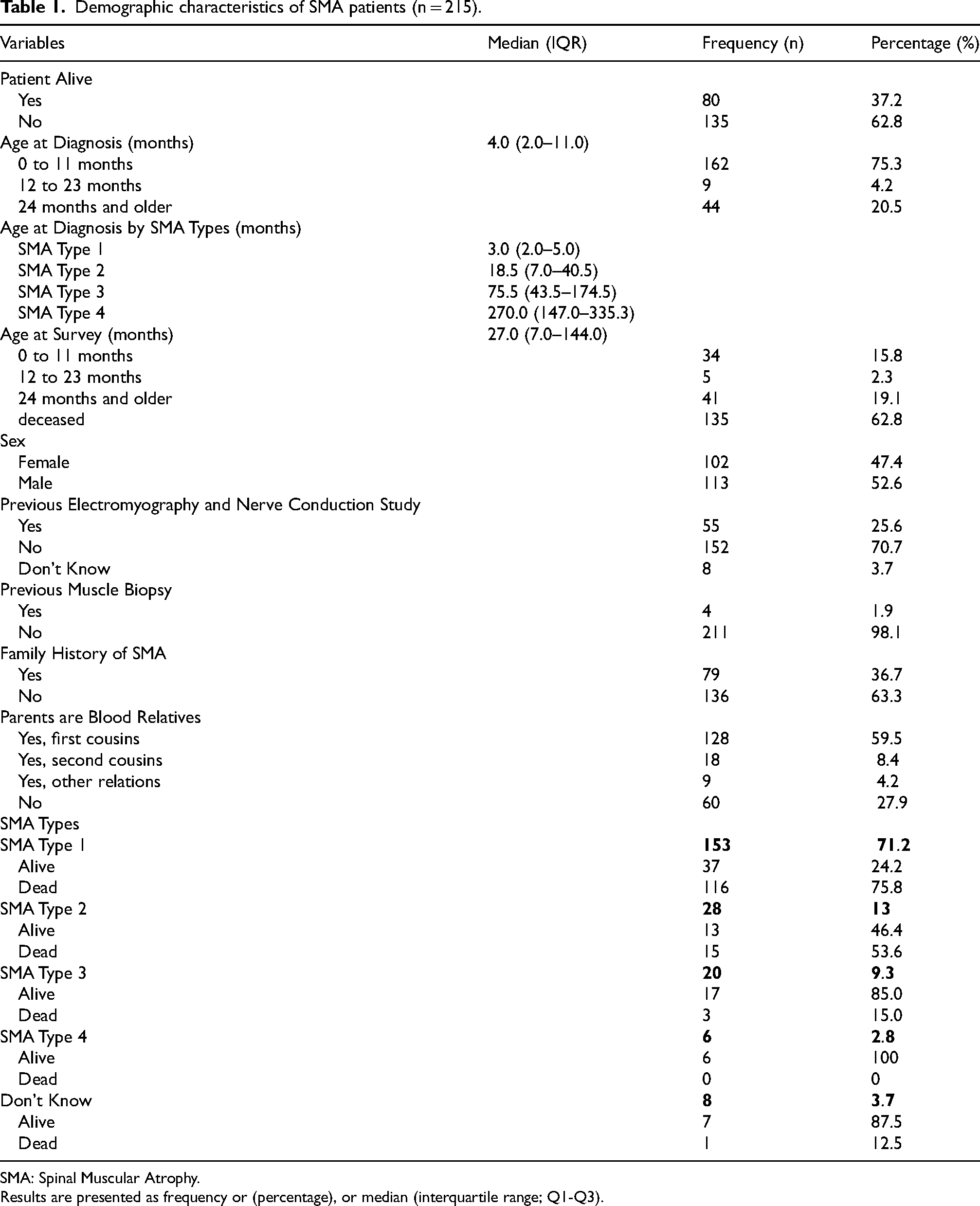
SMA: Spinal Muscular Atrophy.
Results are presented as frequency or (percentage), or median (interquartile range; Q1-Q3).
Our data reveals that 92.6% (n = 199) of study participants were evaluated for SMN2 copies. Amongst patients who were tested for SMN2 copies (n = 199), the majority (84.4%, n = 168) had two SMN2 copies, and 26 (13.1%) had three copies of the SMN2 gene. SMA type 1 patients predominantly carried two SMN2 copies (91.5%, n = 140). Notably, no patients with SMA type 1 had four copies of SMN2 (Table 2).
Frequency of SMN2 copies across SMA types (n = 215).
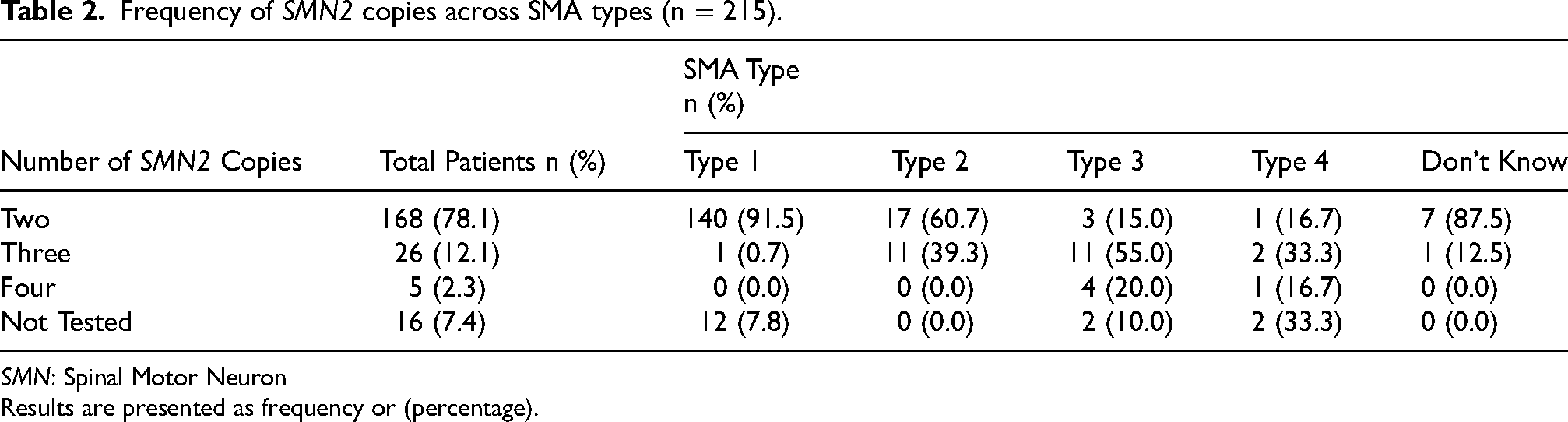
SMN: Spinal Motor Neuron
Results are presented as frequency or (percentage).
Through analysis of the association between medical characteristics and SMA type in patients with known type (n = 207) with chi-square testing, we found SMA types to be significantly associated with the achievement of ever sitting independently at one stage of the participant's life (p < 0.001) and with a previous history of scoliosis surgery (p = 0.041). In patients with known type who were alive at the time of telephonic interview (n = 73), it was found that SMA types were significantly associated with the current ability to independently sit (p < 0.001) and walk (p < 0.001), and usage of a wheelchair (p = 0.0054) (Table 3).
Association between participant medical characteristics and SMA types (n = 207)*.
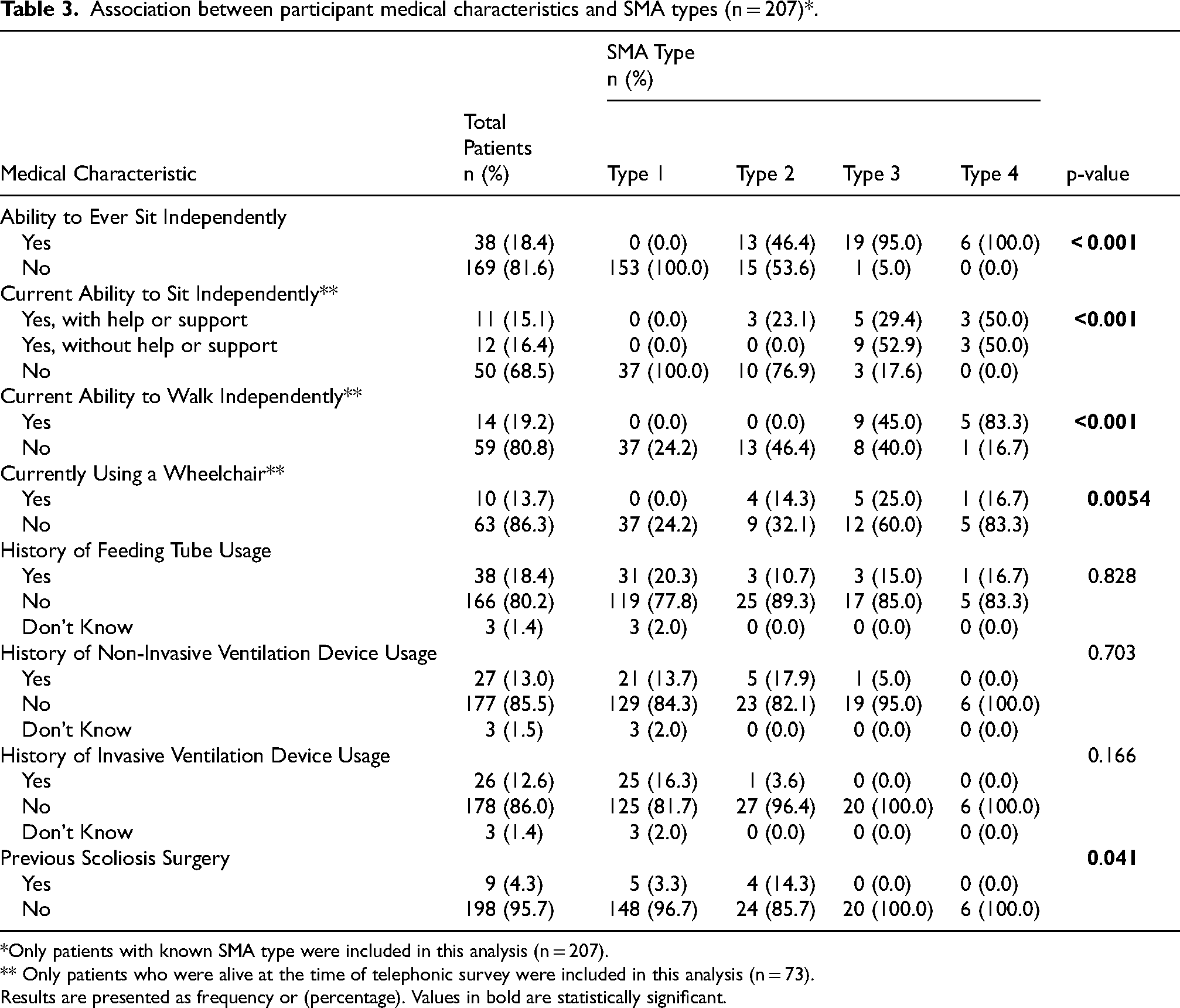
*Only patients with known SMA type were included in this analysis (n = 207).
** Only patients who were alive at the time of telephonic survey were included in this analysis (n = 73).
Results are presented as frequency or (percentage). Values in bold are statistically significant.
In addition, we evaluated the association between medical characteristics of participants and their number of SMN2 copies through chi-square testing. Only patients with known copy numbers were included in this analysis (n = 199). Through analysis of this association, we discovered that the number of SMN2 copies were significantly associated with the ability to ever sit independently (p < 0.001). In patients with known copy numbers who were alive at the time of telephonic interview (n = 76), it was found that SMN2 copy numbers were significantly associated with the ability to sit (p = 0.020) and walk (p = 0.031) (Table 4).
Association between participant medical characteristics and SMN2 copy numbers (n = 199)
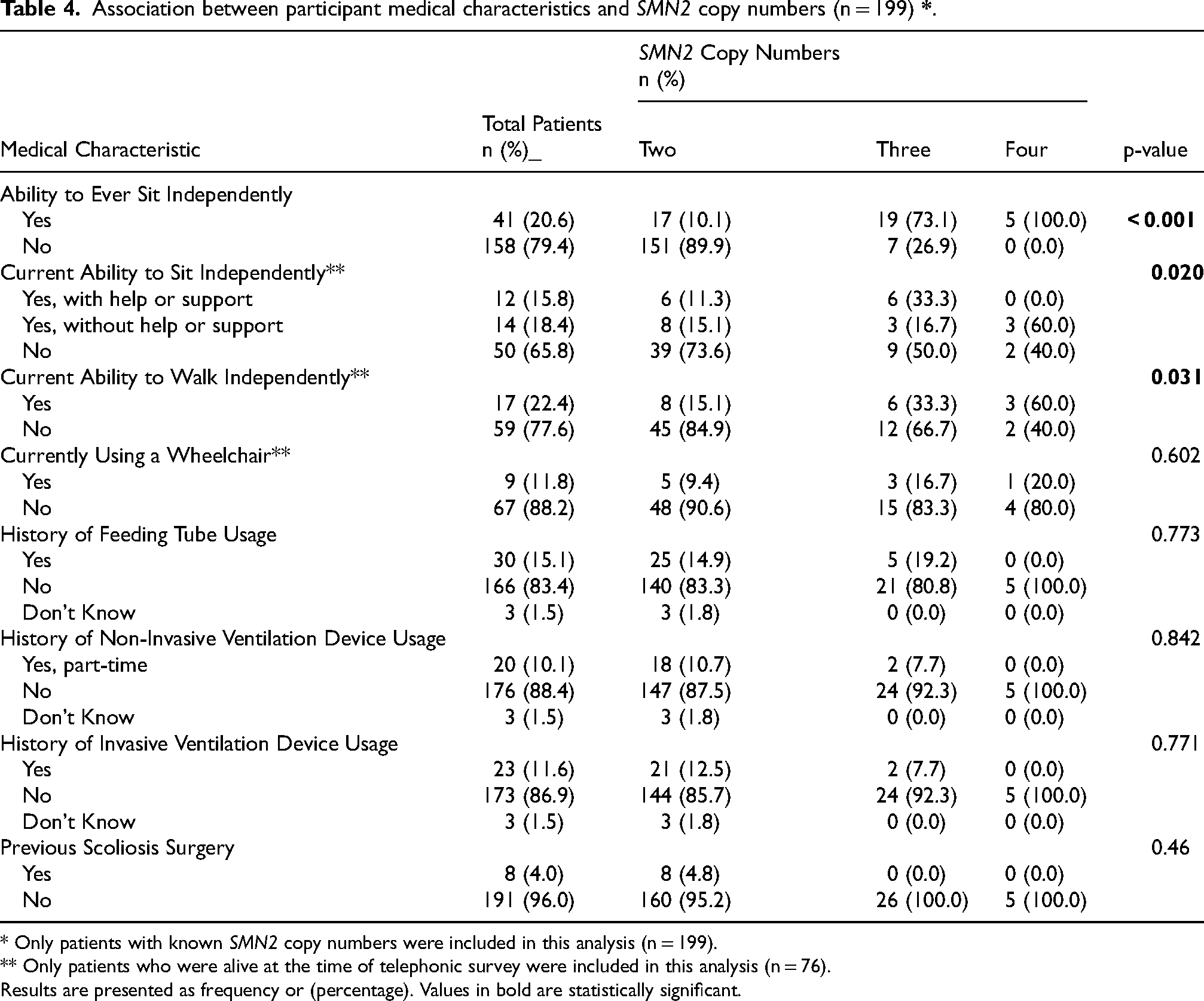
* Only patients with known SMN2 copy numbers were included in this analysis (n = 199).
** Only patients who were alive at the time of telephonic survey were included in this analysis (n = 76).
Results are presented as frequency or (percentage). Values in bold are statistically significant.
In our study population, we found that consanguineous parentage is significantly associated with SMA type (p = 0.049), with SMA type 1 being the most prevalent type amongst children born to consanguineous parents (n = 116, 78.4%). In addition, amongst patients with SMA type 1, we found a significant association between consanguineous parentage and mortality (p = 0.016) (Table 5).
Association between consanguineous parentage and patient variables by SMA types.
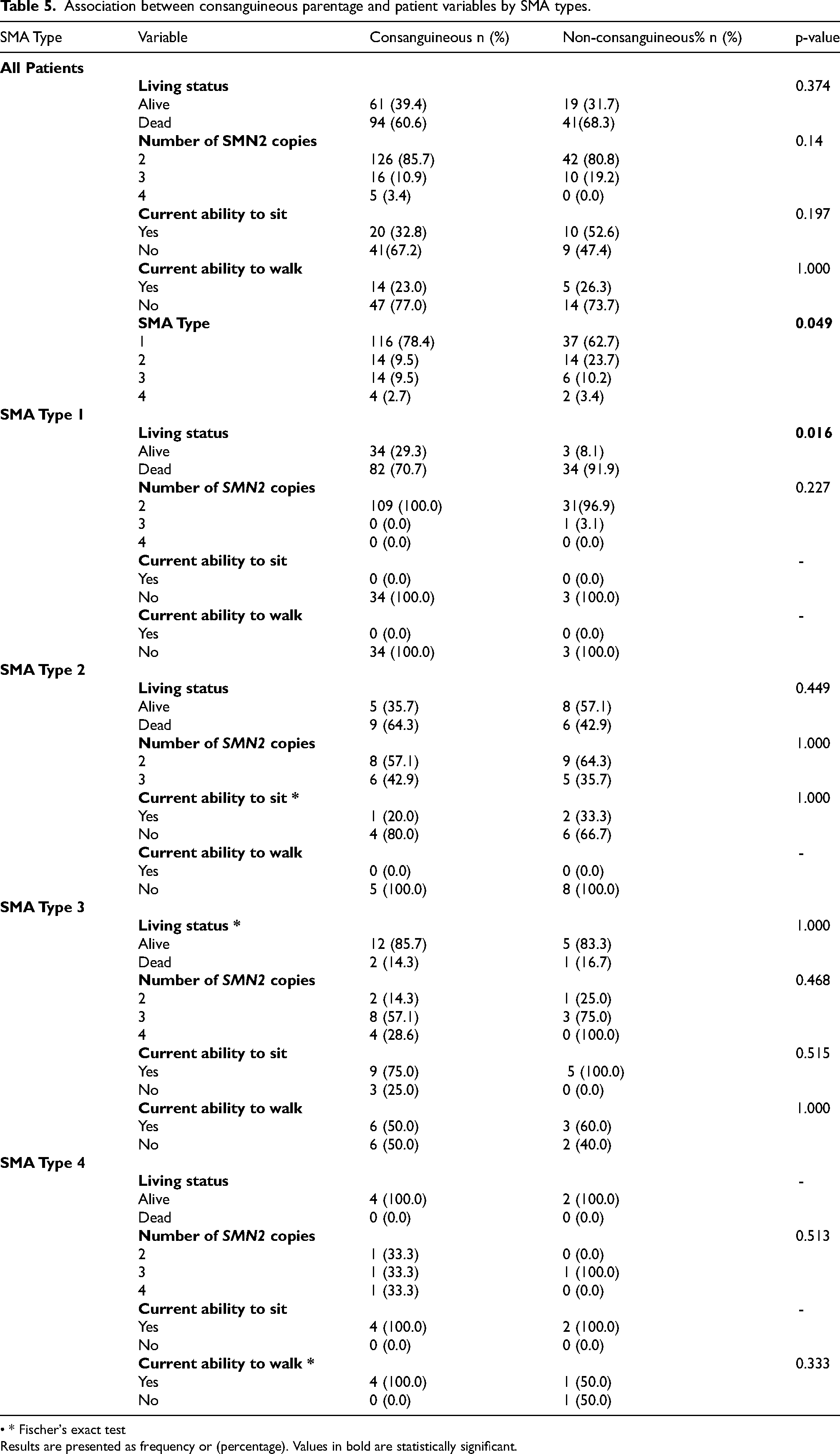
• * Fischer's exact test
Results are presented as frequency or (percentage). Values in bold are statistically significant.
Discussion
In this study, we present descriptive data gathered between 2019 to 2024 on 215 genetically confirmed SMA cases from Pakistan's inaugural neuromuscular disease registry. Our findings reveal that SMA type 1 was the predominant diagnosis amongst our study population, with 71.2% of our study participants being diagnosed with this particular type. These observations are similar with the results of a previous retrospective review conducted in Karachi. 16 Similarly, our findings also align with those from previous data showing SMA type 1 to be the most common form of the disease worldwide, accounting for approximately 60% of all SMA cases globally.5,7,25 However, certain registries have also reported a notable prevalence of SMA type 3.26,27 The higher prevalence of SMA type 1 observed in our study might stem from its’ severe symptoms, notably the respiratory complications of the disease, which prompt parents/guardians to seek early medical attention and genetic testing. However, it is essential to note that many cases of milder SMA, such as types 3 and 4, often remain undiagnosed or have a diagnostic delay, 28 particularly in middle- and low-income countries such as ours, where the high expenses of genetic testing render them inaccessible for a vast majority of the population. Additionally, social norms and cultural beliefs may persuade families to accept these conditions without pursuing comprehensive medical assistance, such as genetic testing and prenatal screening.29–31 This dynamic likely contributes to the observed discrepancy in the underrepresentation of milder SMA types in our study population.
The findings of this study are consistent with the accepted natural history and progression of SMA. For example, it has been previously reported that children with SMA type 1 never achieve the developmental milestone of sitting independently.28,32 In our study, we had no SMA type 1 patient who had achieved this developmental milestone either. It is known that SMA type 1 patients die at a median age of between 7 and 13 months,28,32 similar to the median age at death reported in our study (median: 6 months, interquartile range: 4–9 months). Similarly, prior literature has commented that most children with SMA type 2 achieve the ability to sit independently, but they may subsequently lose this ability, as well as the ability to walk, if they do not receive treatment.32,33 This can be seen in the SMA type 2 patients enrolled in our study as well: 13 had the ability to sit independently earlier on in their lives before their disease became clinically apparent, following which only 3 of these 13 patients retained the ability to sit independently at the time of telephonic survey. In addition, none of the SMA type 2 patients enrolled in our registry were able to walk independently. Both findings can be attributed to the lack of treatment that could have potentially prevented disease progression. Similar phenomena involving the progressive loss of ability to sit independently can be seen in our SMA type 3 patients, highlighting the detrimental and progressive nature of SMA,32–34 which is further compounded by the lack of treatment options available in Pakistan.
The distribution of SMN2 copy numbers amongst SMA types in our study differs somewhat to other globally conducted studies. Prior literature has commented that three copies of SMN2 are common in patients with SMA type 2,4,35,36 with Lusakowska et al. reporting that 82% of their SMA type 2 patients had three copies of SMN2. 26 In contrast, only 11 (39.2%) of SMA type 2 patients in our study had three copies of the SMN2 gene. Studies involving the Dutch and Polish population have reported a high number of patients with SMA type 1 with three copies of SMN2 and a milder disease course.26,37 This differs starkly from our findings, in which only 1 patient out of 153 SMA type 1 patients had three copies of SMN2. This can likely be attributed to the fact that in our population, parents of SMA patients with milder disease course do not opt for genetic testing due to the high associated costs, for reasons discussed above. Furthermore, previous studies conducted in various European countries have observed that 5–6% of patients with SMA type 3 had 2 copies of SMN2;4,26 whereas our study found that 15% of our SMA type 3 patients (n = 3) had two copies of SMN2. This overrepresentation of patients with more severe genotypes can likely be attributed to the various social and economic factors impacting access to healthcare in our Pakistani context, in which milder SMA types have a high likelihood of remaining undiagnosed. It can be hypothesized that patients with higher number of SMN2 copies (and thus, a milder SMA disease course) do not require immediate medical attention or care, and the lack of a national newborn screening program in Pakistan is another reason these patients remain undiagnosed.
Our findings also highlight drastic deficiencies involving respiratory rehabilitation in the treatment of SMA patients. An SMA registry from Spain reported that over 56% of patients were using non-invasive ventilation at the time of the study, 38 compared to 12.6% (n = 27) of the total population of our study participants utilizing this mode of ventilation. It is important to emphasize that the data presented in our study may not accurately reflect the genuine need for ventilation support in our study population due to disparities in intensive care resource allocation across different provinces in Pakistan 39 and among various socioeconomic backgrounds. Limited healthcare insurance coverage in Pakistan, a country in which most citizens pay for medical services out-of-pocket, also impacts access to medical care, particularly for those from the middle and lower socioeconomic classes. As a developing nation, Pakistan encounters considerable challenges in providing sufficient ventilatory support due to resource constraints, such as inadequate equipment and trained personnel to handle critical care cases. 40
We found that patients with SMA type 3 born to consanguineous parents were more likely to have 4 copies of the SMN2 gene, findings consistent with a previous Turkish study. 41 In addition, our study reveals that 59.5% percent of genetically confirmed SMA cases were children born of first-cousin consanguineous unions, while another 8.4% of our participants were born of second-cousin marriages. These figures highlight the role of high consanguinity rates in SMA prevalence. This association has also been previously documented in Middle Eastern countries, 42 where rates of consanguineous marriage are also high.16,17 At 65%, Pakistan has one of the world's highest rates of consanguineous marriages. 21 This high consanguinity rate not only increases the prevalence of SMA patients but also increases carrier frequencies. 7 The high prevalence rate of SMA type 1 reported in our study, especially in patients born to consanguineous parents, has been reported in previous studies conducted within communities with high rates of consanguineous marriages,43–45 and has been attributed to the rarity of the SMN1 mutant gene. 46 These numbers highlight the significance of improving public education and awareness campaigns related to inherited diseases such as SMA, especially as estimated prevalence rates in countries such as Pakistan, with high rates of consanguineous marriage and no national newborn screening program, may be conservative as many patients with severe cases ultimately die before receiving medical care and attention.
Additionally, the high consanguinity rates seen in our population emphasize the importance of making newborn screening for SMA more commonplace. The US Secretary of Health and Human Services Advisory Committee on Heritable Disorders in Newborns and Children has recommended the inclusion of SMA screening to default newborn screening programs, 47 and such screening should particularly focus on cases involving first-cousin marriages and a history of premature infant death due to unknown causes. In countries such as Pakistan, in which there are no newborn screening programs, one possible intervention to reduce SMA prevalence could revolve around educating the population on the associations of consanguineous marriages with hereditary disorders such as SMA. Such an intervention has the potential to reduce the number of SMA carriers, and ultimately, the number of patients with SMA. Another possible intervention to reduce SMA carrier frequency within the Pakistani population involves the initiation of appropriate genetic counseling and offering antenatal genetic testing to parents in a consanguineous union, with family history of SMA, and with family history of premature infant death due to unknown causes. Previous studies have highlighted the importance of physicians located in countries such as ours with high consanguinity rates keeping SMA as a potential differential diagnosis when evaluating patients with clinical presentation similar to SMA to spare patients and their families invasive investigations and delayed diagnoses 48 and to facilitate early initiation of treatment.
Conducting our data through telephonic interviews introduced limitations involving external validity to our study. Our initial plan was to recruit patients presenting to our tertiary care hospital. These patients would have undergone comprehensive assessments including thorough history and physical examinations. However, due to limited patient numbers, we amended our protocol to recruit patients presenting to our clinical laboratory. We then contacted the parents/guardians of patients who tested positive for SMA through telephone calls. With these amendments in patient recruitment, we were able to enroll an increased number of genetically proven SMA patients. As our survey was predominantly conducted via phone, we had to exclude 97 (31.1%) patients whose parents/guardians did not respond or could not be reached despite multiple attempts. Furthermore, in some cases, data could not be collected from parents of recently deceased patients, who were unwilling to discuss details over the phone. It is also worth noting that the study only included children from families who could afford the cost of genetic testing, which leaves out many patients, especially from rural areas. In addition, our study primarily evaluated patients aged younger than 10 years, as only 23 participants (10.7%) were aged 10 years and older at the time of telephonic survey.
As the first Pakistani registry for SMA and DMD, this initiative can pave the way for future research, local clinical trials, and help promote treatment accessibility in the country. Future SMA research in Pakistan should focus on expanding epidemiological studies to include a more diverse and representative population, particularly to those living in rural areas and unable to afford genetic testing. In addition, longitudinal studies to track disease progression and assess the impact of various treatments are also needed with enhanced focus on ensuring availability of rehabilitation services and supportive care for SMA patients. With this data, we hope to advocate for newborn screening programs in the country, particularly for consanguineous families. Ultimately, through this registry, we hope to provide robust data that can be used for clinical trials in Pakistan involving globally-approved treatments.
Footnotes
Abbreviations
Acknowledgements
The authors have no acknowledgments to report.
Ethical publication statement
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Ethics approval statement
This study is consistent with the guidelines set in place by the Institutional Review Boards at Aga Khan University Hospital (5369-Med-ERC-18) and the University of North Carolina at Charlotte (IRB: 18-0444).
Patient consent statement
Enrollment of all participants in the study occurred following the receipt of written informed consent from their parents/guardians. Additionally, verbal consent was obtained during phone interviews, with parents/guardians being informed of their option to withdraw their child from the study at any time.
Statement of artificial intelligence
ChatGPT was used to rephrase and shorten some sentences in the abstract and discussion.
Author contributions
Kulsum Kazi: data curation and acquisition; manuscript writing and editing.
Ahmed A. Arif: conceptualization; funding acquisition; analysis and interpretation of data.
Bisma Aziz: data curation and acquisition; manuscript writing and editing; analysis and interpretation of data; visualization.
Salman Kirmani: data curation and acquisition; manuscript review
Zeeshan Ansar: diagnostic methods; reports; manuscript review.
Asghar Nasir: diagnostic methods; reports; manuscript review.
Shahnaz Hamid Ibrahim: data curation; manuscript review.
Khairunnisa Mukhtiar Ahmed: data curation; manuscript review.
Zahra Hasan: diagnostic methods; reports; manuscript review.
Sara Khan: conceptualization; funding acquisition; supervision; manuscript review
Funding statement
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by NARF - Neurology Awareness & Research Foundation, Pakistan.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The raw dataset used for analysis in this study is available from the corresponding author upon reasonable request.