
Abstract
Background/Objective:
Anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase (anti-HMGCR) myopathy is rare in children. Here, we present a boy with relapsing refractory anti-HMGCR myopathy along with a systematic literature review.
Case Report:
17-year-old boy with five years of muscle weakness, rash, high creatinine kinase (CK) levels, and muscle biopsy compatible with inflammatory myopathy was diagnosed with juvenile dermatomyositis. He was treated with corticosteroids, intravenous immunoglobulin (IVIG), and methotrexate. His muscle weakness improved with this treatment although never completely resolved. CK levels decreased from ∼15000 U/L to ∼3000 U/L. At the age of 15, muscle weakness relapsed after an upper respiratory tract infection; pulse corticosteroid treatment was administered. The re-evaluated muscle biopsy showed a necrotizing pattern and the HMGCR antibody was positive confirming anti-HMGCR myopathy when he was 16. The diagnostic delay was 50 months. Disease activity was monitored by Medical Research Council score, MRI and functional tests. Despite corticosteroids, methotrexate, IVIG, cyclosporine A, and rituximab therapies, muscle weakness improved only slightly during the first three months and remained stable afterwards.
Results of the Literature Search:
We identified 16 articles describing 50 children (76% female) with anti-HMGCR myopathy by reviewing the English literature up to March 1st, 2022. Proximal muscle weakness was the most common clinical symptom (70.8%). Corticosteroids (84.8%), IVIG (58.7%), and methotrexate (56.5%) were preferred in most cases. Complete remission was achieved in nine patients (28.1%).
Conclusion:
Diagnosis and management of children with anti-HMGCR myopathy are challenging. Complete remission is achieved in only one third of these patients. Imaging biomarkers may aid treatment.
Keywords
INTRODUCTION
Idiopathic inflammatory myopathies (IIM) are a group of chronic, autoimmune conditions affecting primarily the muscles. The IIM are classified based on patterns of presentation, age at disease onset, immunohistopathologic features, and response to treatment [1–4]. The major types of IIM include; dermatomyositis [5], polymyositis (PM), inclusion body myositis (IBM), and immune-mediated necrotizing myopathies (IMNM) [2, 6]. IMNM, as the most recently emerging subtype of IIM, are often associated with autoantibodies of anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) and anti-signal recognition particle (SRP) [7, 8]. Anti-SRP and anti-HMGCR myopathies represent 4% and 1% of juvenile IIM patients, respectively [6].
Anti-HMGCR myopathy is usually characterized by progressive proximal muscle weakness prominent in the lower extremities, with the elevation of creatine kinase (CK) [6, 10]. Statins target the same protein with HMGCR antibodies, so this myopathy was first reported in older adult patients after statin exposure [10]. It was later that the disease spectrum expanded to include patients presenting with similar features without statin exposure [11, 12]. Since it is a very rare disease and the number of reported pediatric cases is low, there is no clear consensus on its management. Here, we present a pediatric patient with anti-HMGCR myopathy, who has a relapsing and refractory disease course despite intense immunosuppressive therapies, and analyze the previously published cases.
SEARCH STRATEGY FOR SYSTEMATIC REVIEW
We screened PubMed/MEDLINE and Scopus. We used the keywords of “Anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase myopathy” and “children” and searched all literature from the inceptions of the databases to March 1st, 2022. The search was restricted to English articles. Case reports/series, original research articles, editorials, and review articles about patients with anti-HMGCR myopathy were analyzed. The articles, including data about children with anti-HMGCR myopathy, have been included in the final analysis. Two authors (EDB and SS) independently screened titles, abstracts, and full texts of all relevant articles. Gender, age at disease onset, age at diagnosis, signs and symptoms, laboratory, radiologic, and histopathologic findings, treatments and outcomes of the patients were evaluated in the included studies. Complete remission was defined as a total disappearance of the signs and symptoms of the disease and CK values returning to normal.
DETECTION OF ANTI-HMGCR IGG AND OTHER ANTIBODIES
The serum samples from the patient were screened for anti-HMGCR immunoglobulin G by enzyme-linked immunosorbent assay (ELISA). ELISA plates coated with recombinant HMGCR were incubated with diluted patient samples. Immunoprecipitation of radiolabeled lysates made from mevinolin-treated HeLa cells or full-length HMGCR (created by in-vitro transcription translation) was then performed to determine the presence of HGMCR antibodies. A positive control (high-level human HMGCR antibody serum) was used in the ELISA to confirm its ability to detect a positive result, and a negative control (HMGCR antibody negative serum) to confirm its ability to detect a negative result.
We examined the presence of myositis-specific and myositis-associated antibodies (MSA and MAA) by EUROLINE Autoimmune Inflammatory Myopathies Profile, antinuclear antibody (ANA) by EUROPLUS ANA Mosaics, and extractable nuclear antibody (ENA) by anti-ENA ProfilePlus ELISA and EUROBLOT.
CASE PRESENTATION
Our patient is a 17-year-old male patient, who presented first to a local clinical center with complaints of fever, diffuse nonpruritic maculopapular rash (prominent on the trunk and extremities), abdominal pain, inability to get up from the floor and to stand on one foot for two weeks at the age of 12 years. On his physical examination, the Medical Research Council (MRC) grade was 2/5 for bilateral proximal lower extremities and 3/5 for bilateral proximal upper extremities. The serum CK level was 15000 U/L (normal value ≤ 145 U/L). Findings such as inflammatory cell infiltrates, degeneration and regeneration in some muscle fibers in muscle biopsy suggested inflammatory myopathy, and the patient was diagnosed with juvenile dermatomyositis (JDM). Oral corticosteroid (1 mg/kg/day) and intravenous immunoglobulin (IVIG, 2 g/kg/monthly) were initiated. Subcutaneous methotrexate (MTX, 12.5 mg/m2/weekly) was added to the treatment after a month. The patient’s muscle weakness improved significantly within two months but he did not recover normal muscle strength. MRC grade increased to 4/5 in bilateral proximal lower and upper extremities. And also, CK levels decreased from ∼15000 U/L to ∼3000 U/L.
At the age of 15, he had a marked increase in muscle weakness after an upper respiratory tract infection when he was still receiving MTX and IVIG. His serum CK levels were in the range of 10000–12000 U/L. Magnetic resonance imaging (MRI) of the bilateral thighs revealed diffuse hyperintensity of the proximal levels of the anterior and posterior muscle groups on T2-weighted images. Electromyography (EMG) was reported to show diffuse increased spontaneous activity at rest and during voluntary contraction, polyphasic motor unit action potentials with normal amplitude, duration, and recruitment pattern. These findings were also compatible with myogenic involvement. CAPN3, CEC21, and dystrophin gene mutations were negative. The patient was hospitalized and pulse methylprednisolone (1000 mg/day for three days) and IVIG (1 g/kg/day, one time) were administered. He was then discharged with a treatment plan of 1 mg/kg/day prednisolone, IVIG/monthly, and MTX.
At the age of 16, he was administered to our center for the first time, when he was on prednisolone (10 mg/day), MTX, and IVIG treatments. On physical examination, the patient had a strength of proximal lower and upper limbs below MRC grade 3/5. His neck flexion was MRC grade 4/5. Serum CK level was 8446 U/L (Fig. 1). Alanine aminotransferase (ALT) level was 122 U/L, aspartate aminotransferase (AST) level was 125 U/L, and lactate dehydrogenase (LDH) level was 553 U/L. Inflammatory markers and cardiac enzyme levels were within normal range. ANA was positive at a titer of 1/100, while ENA and MSA/MAA (including anti-SRP) were negative. The previously performed muscle biopsy was reinterpreted. Variation in fiber size, scattered atrophic, degenerating and regenerating fibers along with a few necrotic muscle fibers with occasional phagocytosis, and rare foci of endomysial and perivascular inflammatory infiltration were detected (Fig. 2). The patient’s metabolic tests were completely normal, and with a HMGCR antibody level >550 U/ml (normal value <20), he was diagnosed with anti-HMGCR myopathy. IVIG was discontinued due to lack of efficacy, and cyclosporine A (CsA, 5 mg/kg/day) was added to the treatment.
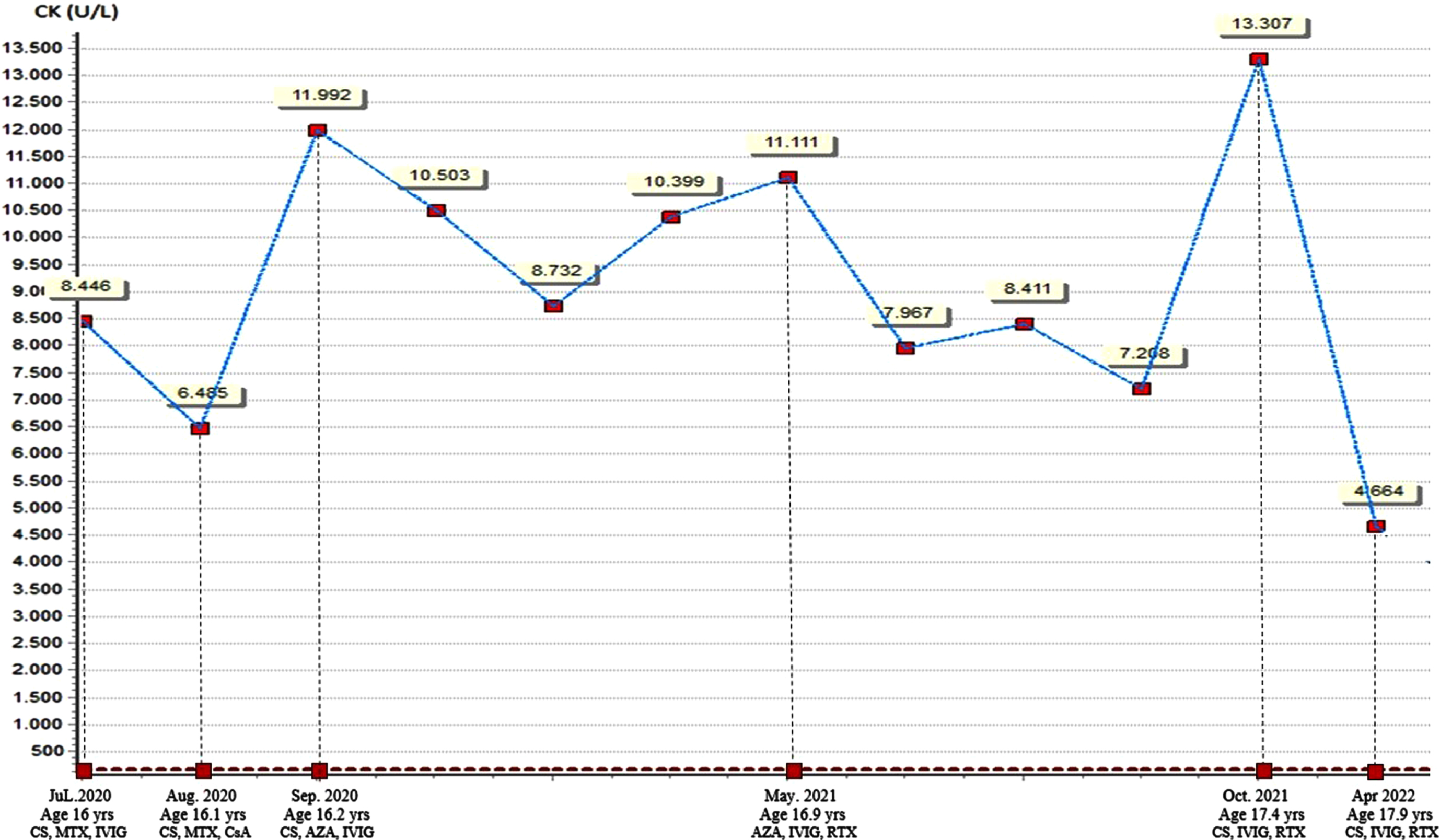
Timeline of the events along with serum creatine kinase levels and treatment regimens. AZA, azathioprine; CK, creatine kinase; CS, corticosteroid; CsA, cyclosporin A; IVIG, intravenous immunoglobulin; MTX, methotrexate; RTX, rituximab.
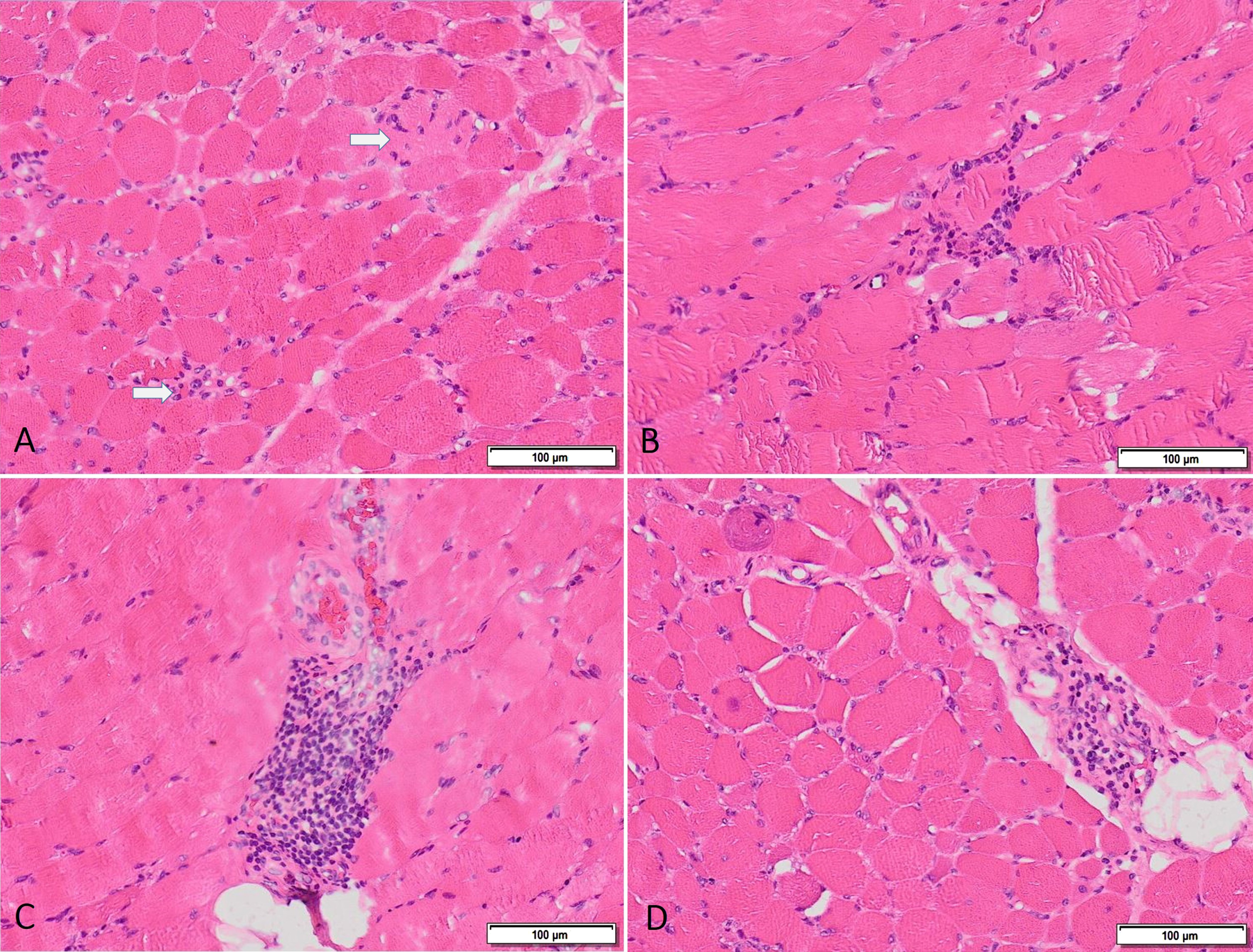
Muscle biopsy shows variation in fiber size, scattered fibers undergoing necrosis and phagocytosis (arrows) (A), foci of endomysial (B) and perimysial (C-D) inflammatory cell infiltration.
Three months later, the serum CK level was still high (11900 U/L) and the muscle weakness did not improve. On whole-body muscle MRI, asymmetrical myositis involvement of bilateral infraspinatus, teres minor and major muscles, pectoralis and serratus anterior, coracobrachialis, triceps and biceps humeri muscles, right paraspinal and iliacus muscles, oblique and transverse muscles of abdomen, quadriceps femoris, bilateral hamstring and gastrocnemius muscles, left tibialis anterior muscles were shown. Involved muscles revealed increased water diffusivity and contrast enhancement without muscle atrophy (Fig. 3). IVIG (1 g/kg/twice a week) was initiated again, and azathioprine (AZA, 2 mg/kg/day) was added to the treatment while MTX and CsA were discontinued. Corticosteroids were tapered and discontinued within three months.
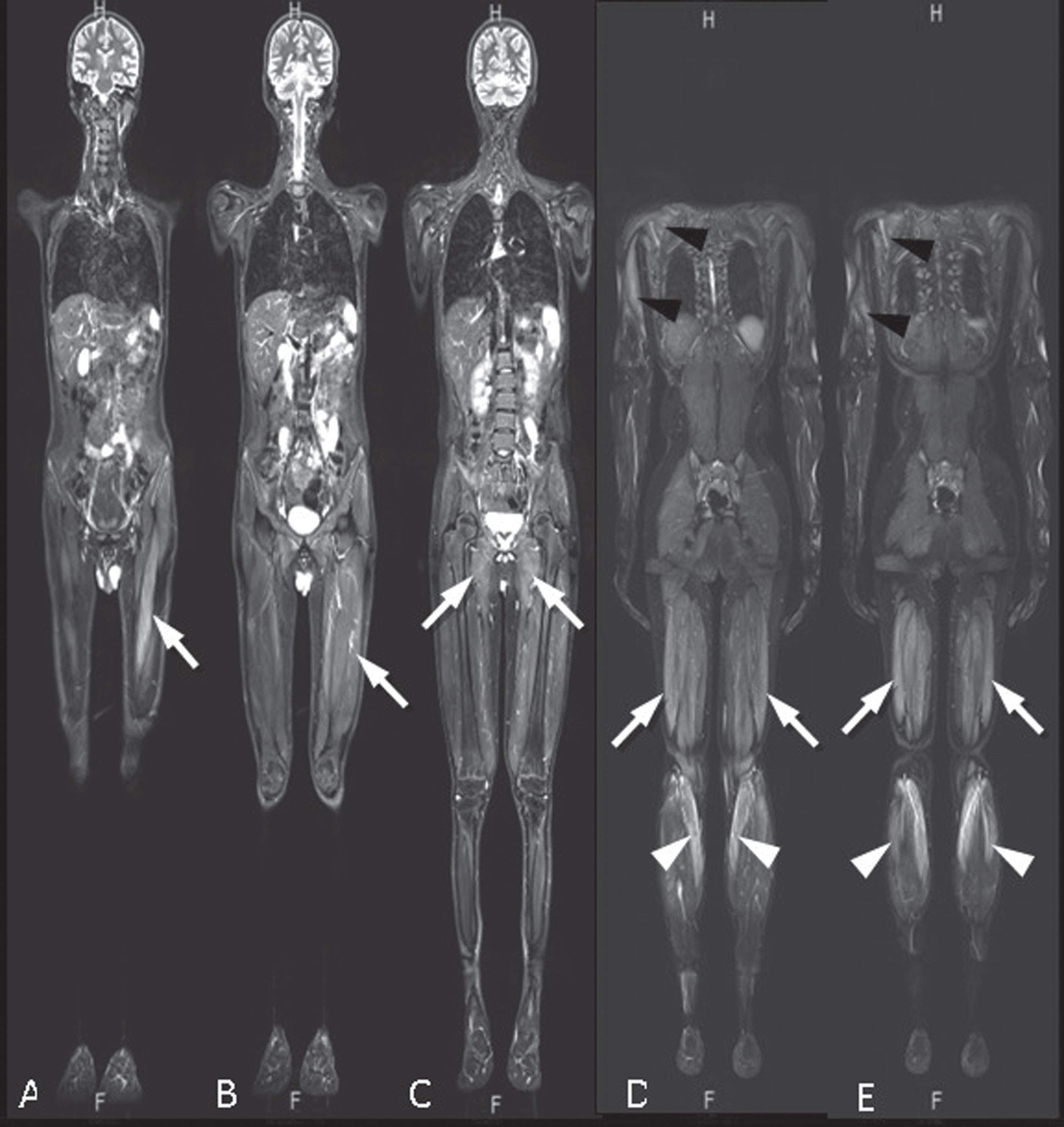
Whole body MRI of the patient revealed myositis as hyperintensity of muscles involving both quadriceps femoris prominent on the left (arrows) (A, B), bilateral adductors (arrows) (C), bilateral triceps, infraspinatus and teres major muscles prominent on the right (black arrowheads) (D) and hamstring (arrows) and gastrocnemius muscles (white arrowheads) (D, E) on coronal plane STIR images. Increased water diffusivity and contrast enhancement of the involved muscles were depicted (not shown).
Despite receiving IVIG and AZA for 6 months, there was no permanent improvement. In addition, the patient specifically indicated that his muscle weakness gradually worsened as the IVIG dose approached, and he got better immediately after receiving IVIG. Therefore, the patient was considered as being IVIG dependent. In addition to IVIG and AZA, the patient was started on rituximab (RTX, 375 mg/m2/week for 4 weeks). After the first RTX cycle, the muscle strength in the upper extremities improved. MRC grade of proximal upper extremity strength increased to 4/5, lower extremity was still 3/5 and neck flexion strength was 4/5. However, the serum CK level was still high (13307 U/L). Thus, low-dose corticosteroid (5 mg/kg/day) was added to the treatment. Follow-up MRI showed generalized mild muscle atrophy and persistence of myositis of the previously involved muscles except thigh adductors which showed mild muscle atrophy without apparent edema and myositis of gluteal muscles accompanying to muscle atrophy (Fig. 4).
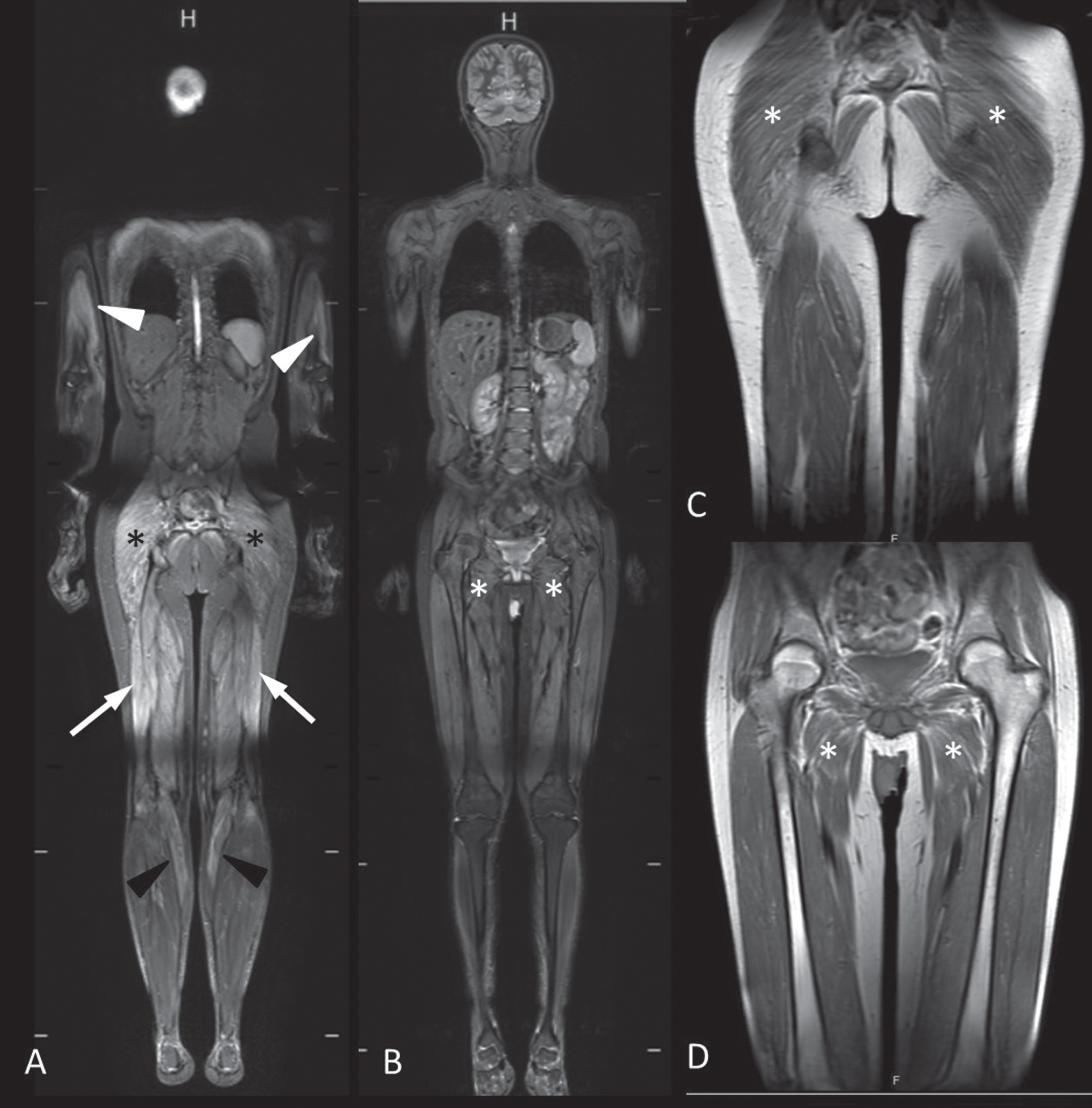
Whole body follow-up MRI of the patient revealed mild myositis of bilateral triceps major muscles (white arrowheads), gluteal (asterisks), hamstring (arrows) and gastrocnemius (black arrowheads) muscles prominent on the right (A), on coronal plane STIR images with mild generalized muscle atrophy characterized by volume loss and hyperintense fatty strikes which are most apparent at gluteal and adductor muscles on coronal plane T1 weighted images (asterisks) (C, D). Interim period images showed myositis of gluteal muscles with atrophy (asterisks) (A, C) with a resolution of myositis and residual atrophic changes of adductor muscles (asterisks) (B, D).
At the patient’s final follow-up (6 months after the RTX course), serum CK values were still high (4664 U/L) while muscle strength was stable compared to the last examination.
CHILDREN WITH ANTI-HMGCR MYOPATHY IN THE LITERATURE
The schematic overview of the literature review process is shown in Fig. 5. We identified 16 articles describing 50 pediatric patients with anti-HMGCR myopathy during the literature search [7, 11–25]. The characteristics of these 50 patients are summarized in Table 1 and are presented in detail in the Supplementary Table 1.
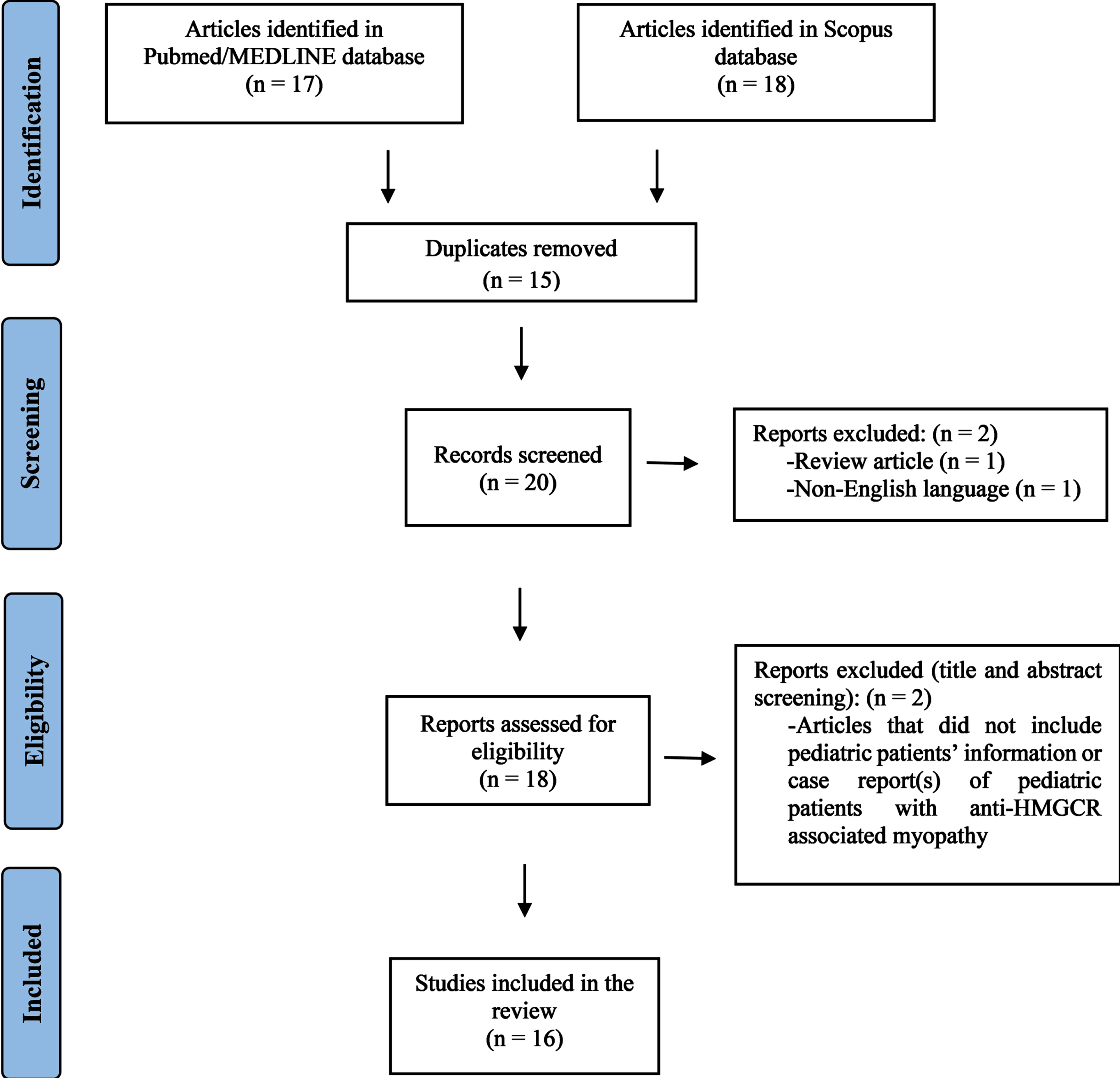
The PRISMA flow diagram of literature screening.
The characteristics of children with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase (anti-HMGCR) myopathy in the literature
Abbreviations: AZA, azathioprine; CK, creatine kinase; CR, complete remission; CS, corticosteroid; CsA, ciclosporin A; CYC, cyclophosphamide; EMG, electromyography; GIS, gastrointestinal system; HCQ, hydroxychloroquine; IFX, infliximab; IVIG, intravenous immunoglobulin; LFM, leflunamide; MAC, membrane attack complex; MHC, major histocompatibility complex; MMF, mycophenolate mofetil; MRI, magnetic resonance imaging; MTX, methotrexate; P62, sequestosome-1; PR, partial remission; RTX, rituximab; TAC, tacrolimus; TNF, tumor necrosis factor.
The median (min-max) age of the pediatric patients with anti-HMGCR myopathy in the literature was 7.5 (0.8–17) years and most of the patients were female (76%).
Proximal muscle weakness was the most common clinical symptom (70.8%) [7, 20–25]. The muscle weakness was more prominent in the lower extremities in 94.2% of patients. Phenotypes varied between muscular dystrophy and inflammatory myopathy. Other musculoskeletal findings of the patients, in order of frequency, were as follows; muscle atrophy (20.8%), distal muscle weakness (18.7%), myalgia (16.6%), neck weakness (8.3%), and asymmetric muscle weakness (4.2%). In some patients (10.4%), other musculoskeletal system symptoms such as joint contracture (n = 1), scapular winging (n = 2), scoliosis (n = 1), and fracture (n = 1) were noted. In addition, 22.9% of the patients had constitutional symptoms such as fatigue (n = 4), fever (n = 3) and/or weight loss (n = 2) [11, 25]. As other extramuscular involvement, some of them had skin findings (37.5%) [11, 22–25], gastrointestinal system symptoms (GIS, 13.9%) [12, 23–25], and cardiopulmonary symptoms (8.3%) [12, 23–25].
The median CK level of the patients was 10198; however, the range (min-max) was quite broad as 435–44002 U/L. The levels of ALT, AST, LDH, and acute phase reactants were mentioned for only one patient in the literature. She had high LDH [1862 U/L (380–640 U/L)], AST [532 U/L (<50 U/L)], and ALT [900 U/L (10–45 U/L)] levels and a normal C-reactive protein (CRP) [24]. Of the 31 patients with muscle MRI, 25 (80.6%) had findings consistent with muscle inflammation/myositis [11, 25]. Myopathic changes were detected in EMG in nine (60%) of the 15 patients [7, 20–23].
The most common muscle biopsy finding was myofiber necrosis (80.8%) [7, 25]. Although the patients’ biopsy findings varied, degeneration or regeneration of muscle fibers (61.7%), inflammation (53.9%), endomysial fibrosis (42.5%), muscular atrophy (21.3%), fiber size variability (19.1%), adipose tissue infiltration (12.7%) and increased internalized nuclei (8.5%) were among the most frequent findings. Membrane attack complex (MAC) deposition (40.4%) [11, 20–23] or major histocompatibility complex-I (MHC-I) expression (57.4%) [11, 21–25] was also observed in some cases (Supplementary Table 1).
In the treatment, corticosteroids (84.8%), IVIG (58.7%), and MTX (56.5%) were preferred in most cases. The most frequently used other immunosuppressive drugs were cyclophosphamide (CYC, 21.7%), RTX (19.5%), AZA (19.5%), mycophenolate mofetil (MMF, 19.5%), and CsA (15.2%). Moreover, biologic agents were also used in seven patients (anti-tumor necrosis factor [TNF] agents in six and abatacept in two) [12, 24]. The median time from the disease onset to initiation of treatment was 12 (2.8–144) months, which was reported for 26 patients.
The median disease duration was 2.6 (0.25–13.1) years. Relapse was reported in 15 (46.4%) of 28 patients. Complete remission was observed in only nine (28.1%), and partial remission in 22 (68.7%) of 33 patients whose outcome was reported. One patient (3.1%) was deceased during follow-up.
DISCUSSION
Anti-HMGCR myopathy is a severe form of IIM [7, 26–28]. In general, these patients tend to require more than one immunosuppressive agent [9]. Some patients (especially adult cases) respond quickly to the treatment and spontaneous recovery can be seen shortly after discontinuation of treatment [20, 29]. On the other hand, the disease shows a resistant course and requires more aggressive treatment in some patients [27].
The majority of IMNM patients had myositis-specific autoantibodies such as anti-SRP or anti-HMGCR [6]. To date, more than 350 patients with anti-SRP and anti-HMGCR myopathies have been reported, allowing us to recognize the clinical phenotypes of patients with IMNM [1, 30]. Frequently, patients with IMNM present with muscle weakness and myalgia [1, 30–32]. On physical examination, proximal muscle weakness is typically prominent in the lower extremities [30–32]. Muscle atrophy is frequently seen, especially in patients with anti-SRP myopathy (∼50%) [33, 34]. Moreover, patients with anti-SRP myopathy have more severe muscle weakness than patients with anti-HMGCR myopathy, and muscle atrophy is more common in these patients [33, 34]. In a study evaluating nine pediatric patients with anti-HMGCR myopathy, which is the largest case series ever reported, proximal muscle weakness was present in eight patients (88.8%), myalgia in two patients (22.2%), skin rash in two patients (22.9%), and constitutional symptoms in three patients (33.3%) [11].
In the literature, dysphagia has also been reported in approximately 30–70% of patients with anti-SRP myopathy and in 25% of patients with anti-HMGCR myopathy, in adults [13, 35]. The rate of dysphagia was 10.8% in children with anti-HMGCR myopathy. Cardiac and pulmonary involvement is very rare in patients with anti-HMGCR myopathy. Only 3% of children with anti-HMGCR myopathy had pulmonary or cardiac involvement. Interstitial lung disease is frequently detected with anti-SRP myopathy and is present in 23–38% of patients [31, 36]. In addition, myocarditis is frequently seen in patients with anti-SRP myopathy [31, 37].
Serum CK levels are typically elevated in both seropositive IMNM [33, 34]. Imaging and histopathology play a very important role in the diagnosis of IMNM. Muscle MRI shows a heterogeneous pattern and asymmetrical involvement with predilection to proximal muscle groups [9]. Whole-body muscle MRI including both fluid sensitive (T2 weighted fat-saturated or STIR) and T1 weighted images can demonstrate both muscle volume and abnormal signal intensity (edema and/or fatty infiltration) [9]. Myositis of muscles is depicted as T2 hyperintensity during active inflammatory phases of the disease. Muscle volume loss with or without fatty infiltration can be demonstrated on T1 weighted images reflecting connective tissue and fatty replacement. In one study, MRI of three patients with anti-HMGCR myopathy showed diffuse and bilateral T2- or STIR-hyperintensity in the thigh muscles [12], and another study showed edematous change of muscles of four patients, atrophic change of muscles in three patients, and fatty infiltration in one patient on the MRI [11]. Imaging follow-up can tailor immunosuppressive treatment strategies. However, although increased STIR signal intensity indicates a room for escalating immunosuppressive therapy, T1 hyperintensity which reflects connective tissue and/or fatty replacement in the muscle is not likely to be reversed by immunosuppressive therapy.
Muscle biopsy findings in anti-HMGCR myopathy are typical of a pauci-immune necrotizing myopathy, showing myofiber degeneration and necrosis with a variable density of regenerating fibers [9]. Perivascular chronic inflammation can occasionally be seen but the lymphocytic invasion of healthy myofibers, as seen in polymyositis, is rarely observed. MHC-I may show increased sarcolemmal staining in non-necrotic fibers and membrane attack complex (MAC) immunostaining (e.g. C5b-9) may stain the sarcolemma with variable intensity as well as endothelial cells occasionally [11, 38]. Similarly, mild endomysial fibrosis, necrosis, and regeneration of muscle fibers and MHC-I expression were detected in all five patients with anti-HMGCR myopathy reported by Hou et al. [14], while lymphocytic infiltrates and MAC deposition were found in four patients. When muscle biopsy is performed after a long period of the disease course, a significant number of atrophic myofibers and fibrosis and adipose tissue replacement can be seen independent of regenerative processes [39]. As these histopathological findings overlap with dystrophic changes, muscle biopsy diagnosis is a challenge and many biopsies have been reported as muscular dystrophy before the recognition of the presence of HMGCR antibodies [18, 40]. The histopathological features usually do not help distinguishing between anti-SRP myopathy, anti-HMGCR myopathy, or seronegative IMNM [41].
Treatment for IMNM is mostly empirical, based on the results of retrospective case series and expert consensus [42]. Corticosteroid monotherapy is insufficient to control the disease in most patients [43, 44]. The majority of patients with IMNM require a second-line agent in addition to corticosteroids within 6 months of starting treatment [45]. MTX has been used as the first choice in most studies to treat IMNM [6, 42]. Other treatment options include IVIG, AZA, MMF, CYC, and RTX [9, 11].
The use of IVIG has been recommended in patients with anti-HMGCR myopathy, especially in resistant cases (if adequate response is not achieved within 6 months of treatment) [42]. There are only a few case reports and case series in the literature describing RTX treatment in refractory cases with anti-HMGCR myopathy [27, 46]. In addition, other biological agents such as anti-TNF agents and abatacept have been used in some refractory patients in the literature [12, 24]. Each of the five cases reported by Kishi et al. [12] required at least one additional immunosuppressive agent in addition to corticosteroids in the treatment. IVIG (n = 4), RTX (n = 2), and other biological agents (n = 2) were also used in some resistant cases. In the future, new therapeutic strategies targeting antibody-producing cells or the complement system may be promising since muscle necrosis in seropositive IMNM is mostly associated with the activation of the classical complement pathway.
Seropositive IMNM has more severe disease in terms of morbidity than seronegative IMNM [47]. Different case series have shown that many patients with IMNM exhibit severe muscle damage [48, 49]. After a long period of illness, it can be assumed that the muscle damage is not completely reversible, even if serum CK levels have been reduced or even normalized with active therapy. The poor outcomes in IMNM patients are probably associated with the extent of muscle damage. The severity of muscle damage depends on the time from the onset of symptoms to the initiation of treatment and the duration of the disease [49].
HMGCR antibody levels are reported to correlate with disease severity in the literature [50]. However, they rarely, if ever, normalize even in patients that appear to have recovered completely [51]. Therefore, the subject of monitoring the antibody level in these patients is still controversial.
Finally, it should be noted that the disease duration is long and the majority of patients require immunosuppressant or immunomodulatory drugs for years after diagnosis, and the side effects and comorbidities of these treatments are also significant concerns [13].
CONCLUSION
Anti-HMGCR myopathy should be kept in mind in juvenile IIM patients especially in the presence of proximal muscle weakness prominent in the lower extremities, markedly elevated serum CK levels, muscle biopsy demonstrating necrotizing features, negative myositis specific antibody profile, muscle MRI signatures, and refractory disease course. Although suggestive, these findings do not always correlate with clinical relapses. On the other hand, the spectrum is expanded with patients presenting with a muscular dystrophy phenotype. Anti-HMGCR myopathy is an uncommon cause of myopathy in children but has treatment. Therefore, it is very important to consider testing for anti-HMGCR autoantibodies in children presenting with subacute, new-onset proximal muscle weakness and/or in the case of a genetically not confirmed myopathy or muscular dystrophy. The outcome is affected by the duration between the disease onset and the initiation of treatment. Thus, early diagnosis and treatment are highly valuable in this disease. However, although spontaneous remission is reported in rare pediatric cases, anti-HMGCR myopathy is not benign and can be relapsing and refractory. There is a need for prospective studies on the long-term efficacy of the currently available drugs, new therapeutic approaches and when to stop escalating immunosuppressive treatments.
FUNDING
No specific funding was received from any bodies in the public, commercial or not-for-profit sectors to carry out the work described in this article.
CONFLICT OF INTEREST
The authors have no conflicts of interest relevant to this article to disclose.
AUTHORS’ CONTRIBUTIONS
All authors contributed to the study conception and design. Data analysis was performed by SS and EDB. All authors contributed to the writing of the manuscript. All authors read and approved the final manuscript.
INFORMED CONSENT
Written informed consent was obtained from the patient for the publication of this case report and the sharing of patient figures.