
Abstract
Background:
There is currently no reliable epidemiological data in the Russian Federation nor data on patient routing and evaluation of the efficacy and feasibility of diagnostic and therapeutic approaches to patients with suspected Duchenne/Becker muscular dystrophy (DMD/BMD).
Objective:
To record and monitor all patients with DMD/BMD in Russia.
Methods:
Observational multicenter prospective & retrospective Registry of patients with DMD/BMD.
Results:
Almost half of the 626 included patients live in the Central and Volga Federal Districts. The most common cause of the disease was large deletions of one or more exons (328 patients, 52.4%). Large duplications were identified in 92 patients (14.7%). Point mutations were identified in 206 patients, 32.9%. Among 448 patients with a known family history, 20% had a first-line relative diagnosed with DMD/BMD. The mean delay in diagnosis (the time from onset to clinically confirmed diagnosis) was 24.3 months.
Conclusions:
These data demonstrate the preliminary results of the Registry and indicate a considerable delay in diagnosis in Russia.
Introduction
Duchenne muscular dystrophy (DMD) is a chronic progressive muscular dystrophy with an X-linked recessive inheritance due to the presence of hemizygous pathogenic variants in the DMD gene. DMD is characterized by early onset (at the age of 2–5 years), progressive muscle weakness, muscle atrophy, gait disturbance, extremely elevated creatine kinase (CK) levels, early disability, and early mortality in patients aged 20 to 40 years. 1 Becker muscular dystrophy (BMD) is an allelic variant with a later adolescent debut and slow progression. 2 The DMD gene encodes dystrophin, a structural cytoplasmic protein part of the dystrophin-associated glycoprotein complex (DAG complex) connecting the muscle fiber cytoskeleton with the surrounding extracellular matrix. 3 The function of the DAG complex is to stabilize the muscle cell membrane and protect it from contraction-induced injury. 4 In the absence of a functional DAG complex, cell membranes are destabilized and destroyed, and, as a result, muscle fiber atrophy occurs.
To date, thousands of different pathogenic variants in the DMD gene have been identified. The most common causes of DMD and BMD are large deletions or duplications involving multiple exons, which account for 70–80% of all known pathogenic variants. In 10–20% of cases, small pathogenic variants are the cause of disease development. 5 The difference in clinical presentation between DMD and BMD is explained by the effect of the variant on the reading frame of the DMD gene's mRNA. In DMD, pathogenic variants lead to a shift in the reading frame and, as a result, to the formation of a premature stop codon. In contrast, BMD is usually caused by changes that do not disrupt the reading frame, allowing the production of a shorter but still partially functional protein. 6
DMD is the most common form of muscular dystrophy in children with an incidence of 1:5000–6000 newborn boys in different regions of the world.7,8 According to the recent meta-analysis of the data of more than 900 million people, 9 the global prevalence of muscular dystrophy is 3.6 per 100 000 people (95 CI 2.8–4.5) with the highest prevalence in Americans at 5.1 per 100 000 people (95 CI 3.4–7.8), followed by Asians with 4.8 per 100 000 people (95% CI 2.7–8.6), Europeans with 3.5 per 100 000 people (95% CI 2.7–4.7) and Africans with 1.7 per 100 000 people (95% CI 1.1–4.5). To date, there are no reliable epidemiological data on the DMD and BMD prevalence in Russia. Given the high disease prevalence and its social significance, many multidisciplinary approaches have been developed for symptomatic, surgical therapy, and rehabilitation of patients with DMD and BMD worldwide.10–12 However, there is no sufficient data on the availability and efficacy of these treatment approaches in patients in Russia. Before the implementation of the current program, there was no national registry on DMD/BMD in Russia, and different centers collected the data in a nonsystematic manner. Thus, data on molecular epidemiology in Russia are limited.
One of the main tasks of maintaining clinical registries is identifying patients who can be administered the targeted therapy. Ataluren (Translarna®) is one of the approved drugs for the treatment of patients with DMD in the United Kingdom, Russia, some countries of the CIS (Commonwealth of Independent States, consists of some of the former Soviet States), and other countries like Chile, Israel, Uruguay, etc.13,14 Ataluren is approved for use in DMD patients with nonsense pathogenic variants starting from two years of age. The use of ataluren leads to the “readthrough” of a premature stop codon and inserting a near-cognate tRNA in its place. 15
Another approach for targeted DMD therapy is the administration of antisense oligonucleotides (ASOs) aimed at modulating splicing and skipping various exons of the DMD mRNA to restore the open reading frame. Several drugs at various stages of clinical studies have demonstrated recovery of dystrophin levels in DMD patient samples and have been approved by the US Food and Drug Administration (FDA): eteplirsen (skipping of exon 51), 16 golodirsen and viltolarsen (skipping of exon 53),17,18 casimersen (skipping of exon 45). 19 With new targeting therapies for DMD, e.g. ASOs, becoming available, it is increasingly important to identify eligible patients.
National registries allow for standardizing patient data collection and solving therapeutic and scientific problems. Maintaining registries may help to identify the main diagnostic barriers, evaluate the long-term disease outcomes, and develop approaches to improve the clinical outcomes and the quality of patient care. Moreover, data from such registries are often used for sampling in clinical trials and identifying patients for targeted therapy.20,21
This study presents the first preliminary results of the National Registry of patients with DMD/BMD in Russia, which includes 626 patients with a confirmed molecular diagnosis. This Registry aims to record and monitor all patients with DMD/BMD in Russia.
Materials and methods
Registry organization
The National Registry of Patients with Progressive Duchenne/Becker Muscular Dystrophy in the Russian Federation (the Registry) was established at the Federal State Budgetary Scientific Institution “Research Center for Medical Genetics named after Academician N.P. Bochkov” (FSBSI N.P. Bochkov RCMG), Moscow, Russia jointly with the Russian Society of Medical Geneticists, and with the financial support of PTC Therapeutics. Aston Consulting provides IT support for the Registry.
Patients
The paper presents the first results of an observational (non-interventional) multicenter cross-sectional study. The Registry includes patients with a confirmed diagnosis of DMD/BMD. The clinical form, when available, was assessed by the referring physician. In other cases, the clinical form was determined based on disease onset: patients with disease onset after 10 years were classified as having Becker muscular dystrophy.22,23 All the study participants (or their legal representatives for minor patients) signed a voluntary informed consent to participate in the study. The study was approved by the Ethics Committee of the Federal State Budgetary Scientific Institution “Research Center for Medical Genetics named after Academician N.P. Bochkov,” Moscow, Russia. All data used in this study have been collected and stored in compliance with Federal Law N 152-FZ of the Russian Federation, which sets out specific requirements for the storage and processing of personal data, including measures to protect it from unauthorized access, disclosure, or destruction. The informed consent form is signed out by the participants at the diagnosis stage, and all participants have the right to withdraw the consent (to date, no participant withdrew the consent for participation).
Method of registration and data collection
The Registry was formed by sending biological material along with a completed questionnaire as part of a free diagnostic program for the diagnosis of DMD/BMD to the FSBSI N.P. Bochkov RCMG. Physicians carried out diagnostic referrals at the patients’ residences. As part of the referral for diagnostics, patients were informed about the possibility of participating in the Registry in the event of a confirmed diagnosis. Along with the referral and the questionnaire containing clinical data, patients referred for tests filled out the informed consent for processing the personal data and consent for medical genetic counseling and genetic testing. Participation in the Registry is voluntary and may be withdrawn by a legal representative at any time, provided that the withdrawal is made in writing.
Collected data characteristics
The collection of clinical data for patients referred for DNA diagnostics and participation in the Registry took place from June 2021 to May 2022, in conjunction with the collection of retrospective data. The following was indicated when referring patients for diagnostics: the presumptive clinical diagnosis, family history, age of onset, the ability to walk independently, and the main laboratory parameters, namely the level of CK, alanine aminotransferase (ALT), aspartate aminotransferase (AST), and lactate dehydrogenase (LDH) in the blood serum. From January 2022, it was decided to expand the range of collected parameters, including detailed clinical data, laboratory parameters, muscle biopsy data, the results of instrumental diagnostic methods, treatment methods, rehabilitation, and molecular genetic data. Upon confirmation of the diagnosis of DMD/BMD and the presence of a completed informed consent, the expert team at the FSBSI N.P. Bochkov RCMG decided to include the patient in the Registry. The criteria for the incorporation of a patient in the Registry were an established clinical diagnosis of DMD or BMD in male or female patients of any age, the presence of an informed consent signed by the patient or his/her legal representative, and consent for the processing of the personal data, the identification of pathogenic or likely pathogenic variant in the DMD gene. The prospective nature of the Registry will involve follow-up of patients in the Registry at the place of residence with recording by experts the up-to-date data on the disease progression and treatment methods. Classification is clinician-reported and based on the age of onset and clinical manifestation; the questionnaires are filled out by clinicians.
DNA-diagnostics
Molecular genetic diagnostics were performed at the DNA-Diagnostics Laboratory of FSBSI RCMG. All 79 exons of the DMD gene were analysed for deletions and duplications using MLPA. For MLPA-negative samples, a targeted NGS gene panel was applied, followed by Sanger sequencing for confirmation. Variant annotation followed HGVS nomenclature and ACMG guidelines. The OMIM database, the HGMD® database, version 2021.4, the specialized LOVD database (https://databases.lovd.nl/shared/genes/DMD), and the ACMG guidelines 24 were used to assess the clinical significance of the identified variants. Detailed information on DNA extraction protocols, sequencing platforms, gene panel composition, bioinformatic pipelines, and reference databases used for variant interpretation is provided in the Supplementary Material.
X chromosome inactivation
According to Allen RC et al. 1992, 25 the X chromosome inactivation pattern was investigated in blood. For this purpose, we analyzed the methylation pattern of the polymorphic (CAG) repeat in exon 1 of the AR gene on the X chromosome using quantitative fluorescent methylation-specific PCR (QF-PCR) followed by fragment analysis on an ABI3130xl genetic analyzer in blood samples (Applied Biosystems, United States). The X chromosome inactivation ratio (XCI ratio) was calculated according to the formula proposed by Bolduc et al. 26
Other cohorts
To assess the differences between our patient cohort and the global ones, we compared their clinical data to the global spectrum of DMD using the TREAT-NMD DMD Global Database. 27 By comparing our patient data to a global cohort, we aimed to gain a better understanding of the unique features and characteristics of DMD in our patient population.
Statistical analysis
The analysis of differences between the Russian cohort and other published cohorts in terms of the spectrum of pathogenic variants was assessed using the Fisher exact test. Differences were considered statistically significant at p < 0.05. When calculating mean and median values by age, the value of biochemical analyses in parentheses indicates the interquartile range.
Results
Basic characteristics
From June 2021 to May 2022, 427 patients underwent molecular analysis as a part of the free diagnostic program (Table 1). In 309 patients (72.4%), a pathogenic/likely pathogenic variant was detected in the DMD gene. In 66 patients (15.4%), there were no findings, and 52 patients (12.2%) carried pathogenic variants in other genes associated with limb-girdle muscular dystrophies. The other 199 patients were recruited retrospectively using data from the DNA diagnostic laboratory of the RCMG. As of June 2022, there are 626 patients with an established clinical and molecular diagnosis of DMD/BMD in the Registry. Of the 626 patients, 560 (89.45%) were reported to have DMD and 57 (9.1%) BMD. In 9 patients (1.45%), there were no sufficient clinical data to classify the patients as having DMD or BMD.
Baseline characteristics.
Demographically, the Registry includes patients from all eight federal districts of the Russian Federation (Figure 1(a), Table 1). Almost half of the patients in the Registry live in the Central and Volga Federal Districts, which account for 25.1% and 24.3%, respectively. The smallest number of patients in the Registry is represented by patients from the Far Eastern Federal District (5%). The most represented regions in the Registry are Moscow (74 patients), Samara (38 patients), and Sverdlovsk regions (30 patients).
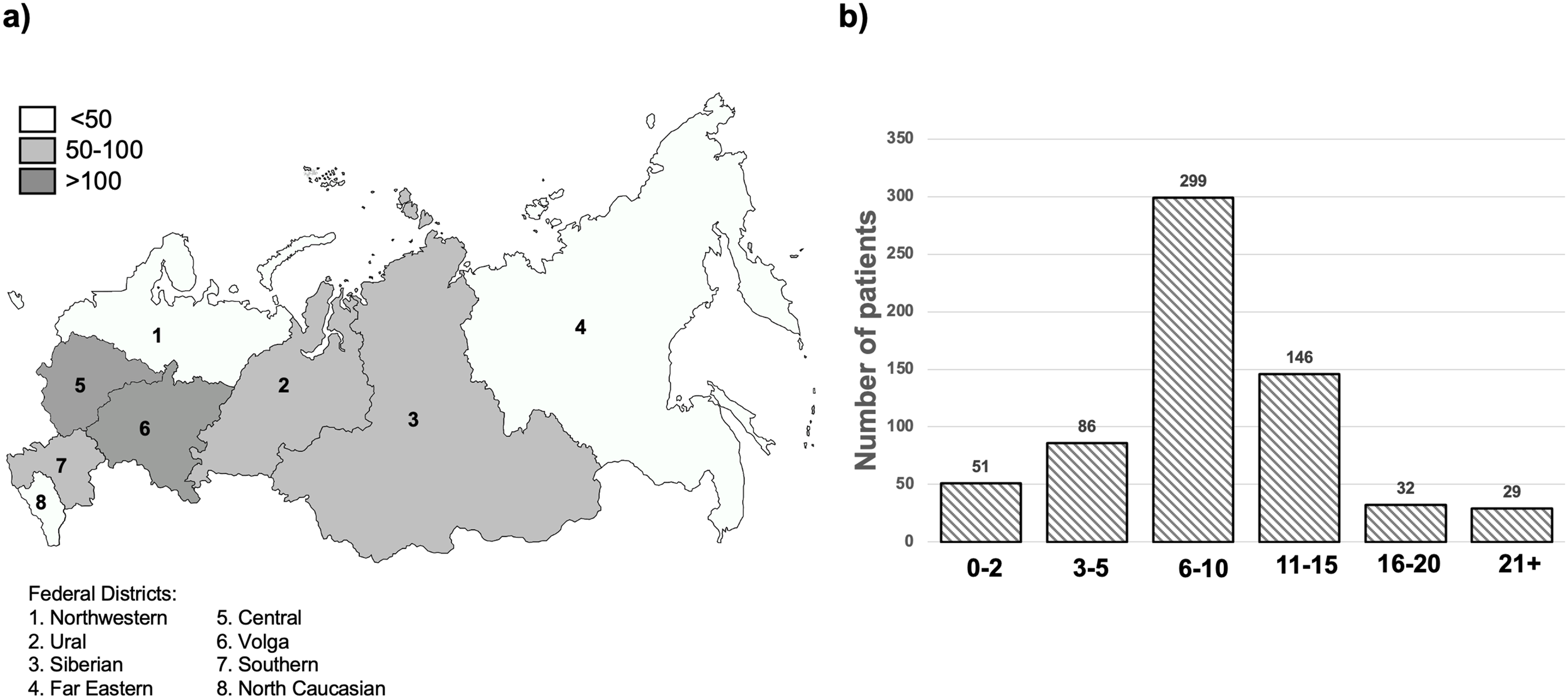
Patient distribution in the DMD/BMD registry: (a) based on the federal district; (b) based on age.
Most of the patients in the Registry are under 16 years of age. The largest number of patients are children (6–10 years old) and early adolescents (11–15 years old) (Figure 1(b), Table 1). The median age of patients in the Registry is 9 years (6–12). The patient's age range is 1–54 years. There are 625 male and one female patient in the Registry.
Preliminary analysis suggests that there is a delay in DMD diagnosis in Russia compared to other countries. According to our data, the mean delay in diagnosis (time between the onset and the clinical diagnosis) in the Russian sample was 24.3 months
Molecular genetic characteristics
Among the 626 patients in the Registry, the most common type of pathogenic variant was large deletions involving one or more exons (328 patients, 52.4%). Large duplications were identified in 92 patients (14.7%). Small pathogenic variants were identified in 206 patients (32.9%, Figure 2(a)). Among these, nonsense variants were most frequently identified in 104 patients, followed by small deletions identified in 47 patients, and variants affecting splicing sites in 27 cases.
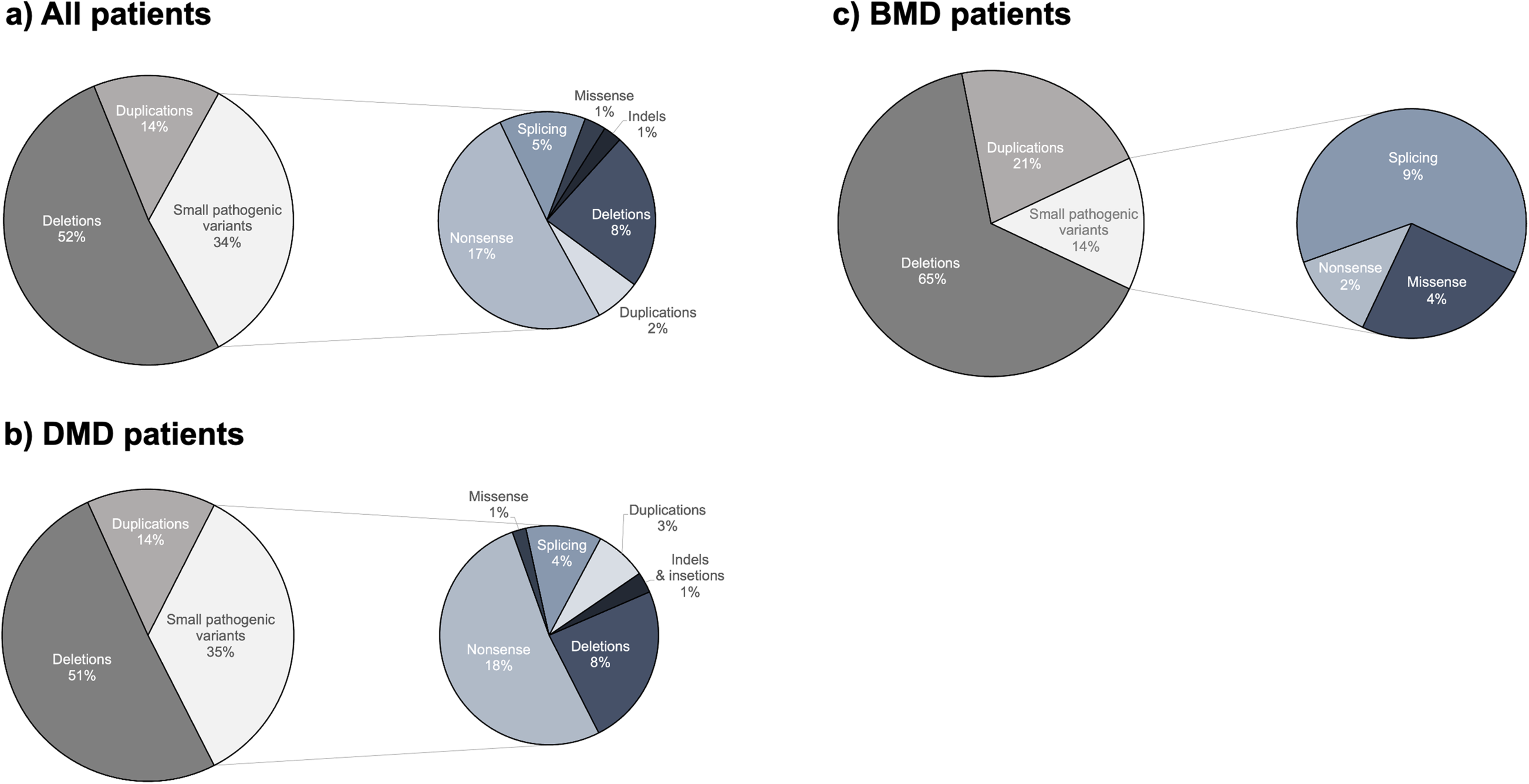
Mutational spectrum of patients in the DMD/BMD registry.
In the DMD patient group, the distribution of pathogenic variants was nearly identical, with large deletions found in 284 patients (50.7%) of cases, large duplications in 80 patients (14.3%), and point pathogenic variants in 196 patients (35%) (Figure 2(b)). In the large deletions group, 83.45% of deletions led to a frameshift, whereas 11.27% were predicted to lead to in-frame deletion. In 15 patients (5.28%), deletions involved the first and/or last exons of the DMD gene with an effect that is difficult to predict. In the large duplications group, 18 patients (22.5%) were found to carry in-frame duplications. When analyzing the point pathogenic variants group, the most frequent were again nonsense variants, accounting for 52%, followed by deletions (24.1%), splice site variants (11.22%), duplications (7.68%), indels (3%), and missense variants (2%). Only four deletions and one indel led to an in-frame change. Analysis of the distribution of nonsense variants showed that the unique 90 nonsense substitutions were found in exons 6–75, similar to other reports, when the N-terminus and C-terminus of the dystrophin protein were depleted with nonsense variants in DMD patients. 28
In the much smaller BMD patients’ group, we observed a distinct mutational spectrum with large deletions accounting for 37 patients (65%) and large duplications in 12 patients (21%, Figure 2(c)); in BMD patients, point pathogenic variants were found only in 8 cases (14% compared with 35% in DMD patients). In BMD patients, all deletions and duplications were in-frame, except for one case with a nonsense variant in exon 31. Exon 31, an in-frame exon was previously found to be enriched with nonsense variants in BMD patients. 28
The comparative analysis of the pathogenic variant spectrum in our Registry and the global spectrum according to the TREAT-NMD DMD Global Database 27 showed (Table 2) that the proportion of the patients with large deletions was significantly lower in our cohort compared to the global dataset compared to the global data (52.4% vs. 68%, p < 0.00001), large duplications occur more frequently (14.7% vs. 11%, p = 0.0068), and the proportion of the patients with point pathogenic variants was significantly higher (32.9% vs. 10.6%, p < 0.00001). These differences remain statistically significant when adjusted for multiple comparisons, which means that the patient cohort with DMD/BMD in Russia differs significantly from the global one. When comparing the DMD patient group from the Registry with the TREAT-NMD DMD Global Database, we observed the same results as before, except that small insertions were found to be significantly less common in our cohort (p < 0.001, Supplementary Table 1).
Comparison of the registry patients and the global TREAT-NMD cohort.
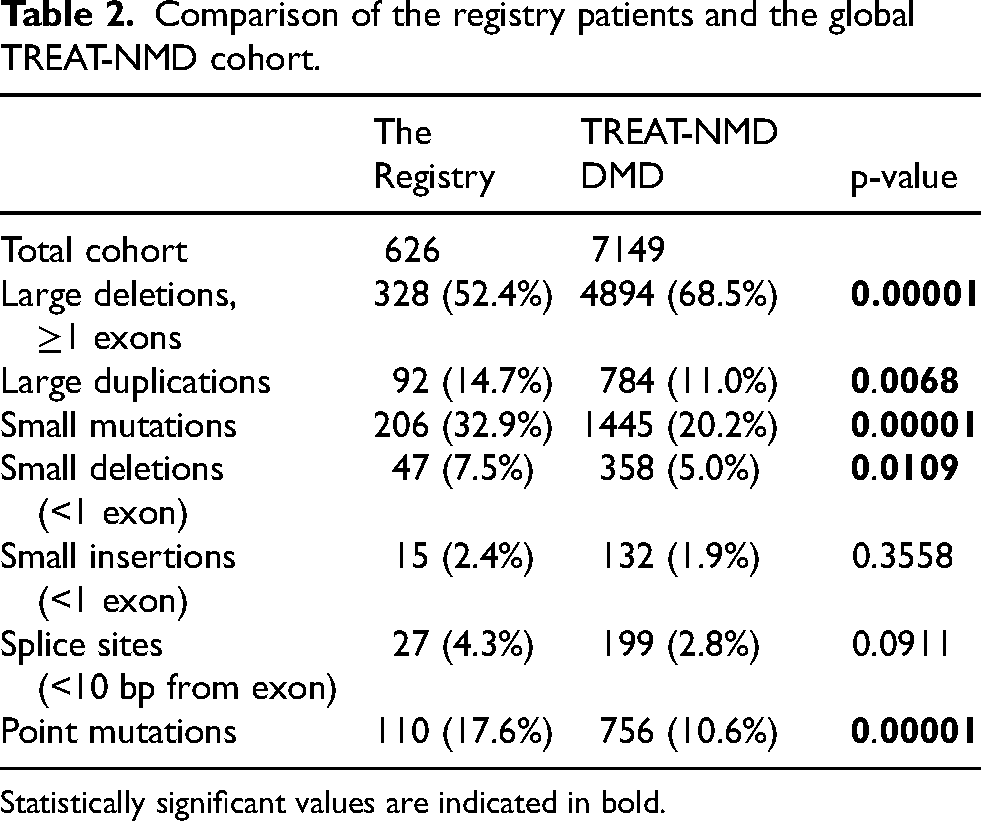
Statistically significant values are indicated in bold.
Clinical profile
Among 448 patients with a known family history, 80% were sporadic, and 20% had a first-line relative diagnosed with DMD/BMD. Among all patients, 59.5% remain ambulatory at the time of entry into the Registry. The data on the disease progression stage are available for 232 patients. Among them, at the time of adding to the Registry, 13 patients were diagnosed at a preclinical stage (Figure 3). Most patients were identified at the ambulatory stage (110 at the early and 69 at the late ambulatory stage); 40 patients were identified at the non-ambulatory stage, 3 of which were at the late non-ambulatory stage. Among the patients diagnosed at the preclinical stage, 6 cases were familial, and the diagnosis was established after the diagnosis of the older sibling. In the remaining 7 cases, the diagnosis was suspected due to the accidental detection of a dramatic serum CK elevation. Among patients with the available data on age of onset, the mean and median ages were 48.5 and 38 months, respectively. The most common symptoms at the debut were muscle weakness, gait disturbance, and frequent falls (the questionnaire may be found in the Supplementary File). Most often, the DMD/BMD diagnosis was first suspected by neurologists (78% of all cases). Other physicians who suspected the DMD/MDD diagnosis were geneticists (12%), as well as pediatricians, orthopedists, hepatologists, gastroenterologists, infectious disease specialists, and cardiologists in single cases.
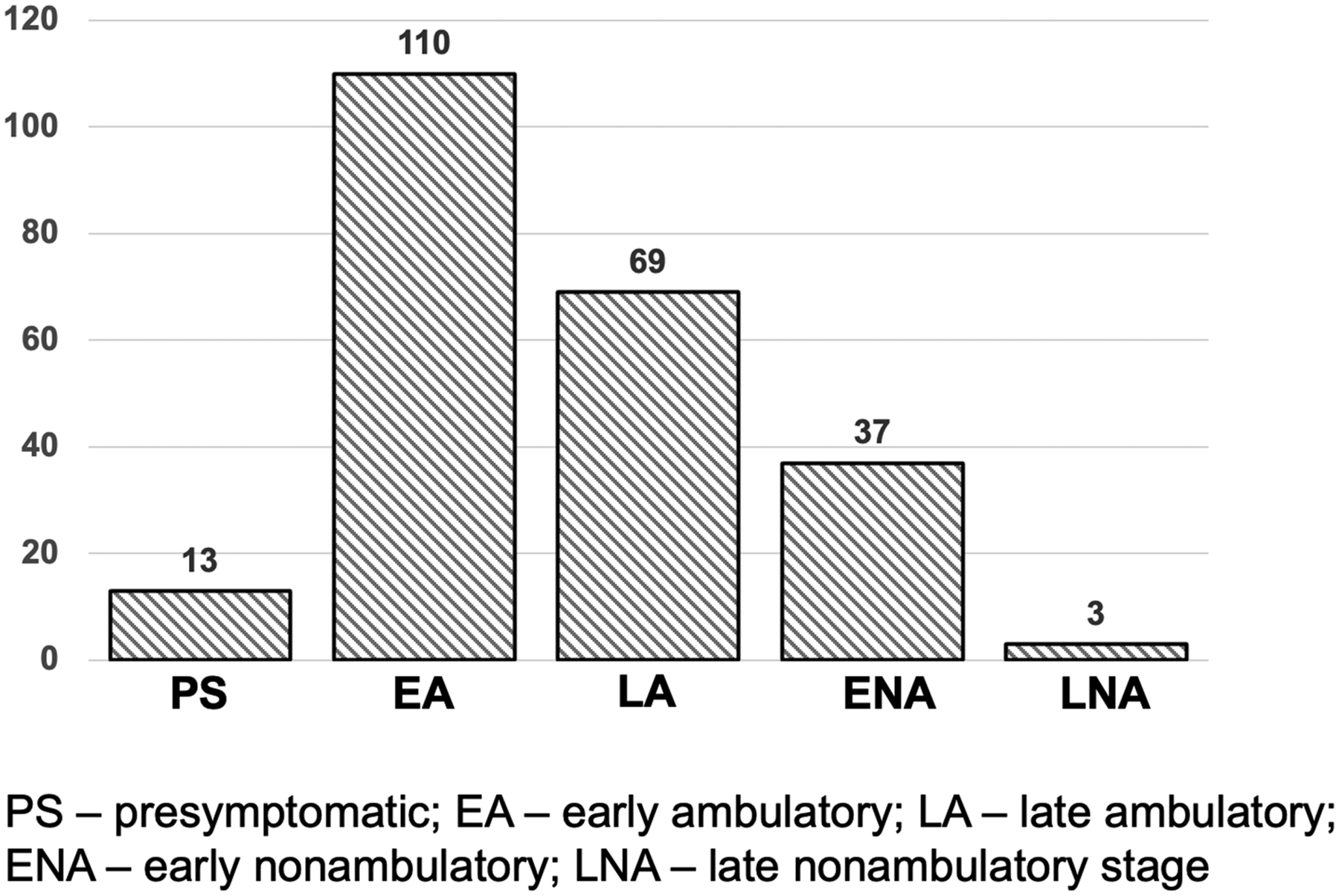
Distribution of clinical stages of patients in the DMD/BMD registry. Early ambulatory stage – patients are ambulatory and can climb up stairs. Late ambulatory stage – patients are ambulatory, but due to progressive muscle weakness are unable to climb up stairs and stand up from the ground. Early nonambulatory stage – Patients are mostly nonambulatory with severely reduced capacity for independent walking. Late nonambulatory stage – Patients are completely nonambulatory with reduced upper limb functioning, difficulties with maintaining body position.
Among the laboratory parameters, 100% showed an elevation in the level of CK. The median value was 8762 U/L (4724–15122) with a spread of values from 893–68,500 U/L. The elevations in serum ALT, AST, and LDH are common early biomarkers in patients with DMD. 29 An elevation in these biochemical parameters can mislead physicians about the presence of liver pathology in patients. In our sample, the majority of patients showed an increase in all 3 parameters. The median values of ALT, AST, and LDH were 259 U/L (136–388), 194 U/L (120–304), and 1166 U/L (723–1530), respectively.
A female patient aged 6 years at the time of examination is one of the interesting clinical findings. The patient was consulted by a geneticist due to muscle weakness, gait disturbance, and difficulty climbing stairs. The first complaints of gait disturbance appeared at the age of 2 years. During hospitalization, the CK level elevation to 5107 U/L and an increase in serum ALT, AST, and LDH were revealed. Examination showed hypertrophy of calf muscles, contractures of the ankles, and tendon areflexia of the lower extremities, and positive Gowers’ sign. The female patient was diagnosed with limb-girdle muscular dystrophy (LGMD).
When searching for pathogenic variants in 15 genes responsible for the LGMD development, a nonsense variant in the DMD gene previously described as pathogenic 30 was identified in the heterozygous state—NM_004006.2:c.9380C > G (p.Ser3127Ter). Segregation analysis determined that the variant was inherited from the proband's healthy mother. Considering the patient's gender, an assumption was made about the presence of an unbalanced X-inactivation. The analysis of the X-inactivation pattern revealed a non-equilibrium inactivation in the ratio of 95–5%. It was found that the allele of paternal origin is predominantly methylated. Therefore, the diagnosis of DMD was established based on the combination of clinical and molecular genetic data of the female patient and the lack of genetic findings in other LGMD-associated genes.
Possibilities of using the targeted therapy
Ataluren is approved for use in DMD patients with nonsense pathogenic variants starting from two years of age. Among the 104 patients in the Registry with nonsense pathogenic variants, 103 patients meet these criteria, which is 16.4% of all patients in the Registry. When analyzing the proportion of patients amenable to exon skipping therapy, we observed that 38 patients (6.1%) have a suitable genotype for exon 51 skipping; 24 patients amenable to exon 53 skipping; 32 patients amenable to exon 45 skipping, totaling 94 patients (15%) that could be treated with exon-skipping therapy. Overall, targeted therapy can be administered to a total of 197 patients from the DMD/MDD Registry (31.5%).
Discussion
Given the lack of systematically collected data on patients with DMD and BMD in Russia, creating a National Registry is of great application and scientific interest. The cross-sectional nature of the study makes it possible to evaluate both the mutational spectrum of patients diagnosed in different years and identify the main difficulties that patients face in diagnosing and prescribing treatment, estimate the effectiveness of these therapeutic measures over time, and assess the routing of patients from onset to long-term disease outcomes. Incorporation of a large number of patients into the Registry will allow us to estimate the real incidence of DMD and BMD not only in the country as a whole but in individual regions in the future, which supports the maintenance of the Registry and is one of the strengths of the current study.
Currently, in Russia, a diagnostic algorithm is applied to a suspected DMD/BMD diagnosis, including a biochemical blood test, genetic, morphological (immunocytochemical staining), and instrumental (ultrasound, MRI) methods. The gold standard for diagnosing DMD is genetic testing and muscle biopsy. However, the availability of the tests in Russia might differ from region to region.
The collection of detailed information about the clinical picture and therapeutic measures with case follow-up is an essential component of the Registry and an important aspect in assessing the efficacy and feasibility of diagnostic and therapeutic approaches used in Russia and their compliance with international standards. It is known that administering glucocorticosteroids in patients with DMD and BMD significantly increases muscle strength, improves motor functions, slows down the disease progression, and prolongs the outpatient stage of the disease. 31 While it is challenging to determine how widely these drugs are prescribed, patient registries help address this issue by providing large-scale, real-world data on prescribing patterns, as demonstrated by other registries. 32 Such data can inform clinical guidelines and improve treatment standardization. The importance of nationwide registries becomes even more evident when new targeted treatments for patients with DMD, such as ataluren and ASO-based drugs, become available. As our analysis showed, today, more than 30% of patients in the Registry can potentially be admitted to targeted therapy.
An interesting observation is the biased mutational spectrum of our sample relative to global data. Our sample includes significantly fewer large deletions, more large duplications, and point pathogenic variants. For instance, in the current study, among patients with the available data on age of onset, the mean and median ages were 48.5 and 38 months, respectively, consistent with literature data. 33 It was shown that more than half of the patients had large deletions (≥1 exon), significantly lower than in the TREAT-NMD DMD cohort. However, many more patients from the Registry had point pathogenic variants. Differences in the proportion of large deletions and point pathogenic variants persist when comparing our cohort with individual national cohorts, such as Japanese, 34 Kuwaiti, 35 and Chinese. 36
The results suggest that there is a delay in DMD diagnosis in Russia compared to other countries, such as the UK 37 and the US, 38 with a similar onset age (72.9 months versus 51.7 and 58.8, respectively). The mean delay in diagnosis in Russia was 24.3 months. These data may indicate a lack of awareness among physicians about DMD/BMD.
Considering that only 1/3 of pathogenic variants in the DMD gene arise de novo, and in other cases, the pathogenic variant is inherited from mothers,39,40 one explanation may be a mixed mutational spectrum in carrier mothers and a higher carrier state rate in our population. A systematic study of mothers of patients from the Registry will allow us to confirm or refute this assumption. The sample bias relative to the world data can also be due to the limitations and peculiarities of the Registry formation. The Patient Registry is mainly formed by physicians’ referrals from other regions for diagnostics at the FSBSI N.P. Bochkov RCMG. The bias towards a smaller number of large deletions may be due to the fact that some of the patients referred for diagnostics at the FSBSI N.P. Bochkov RCMG are analyzed for frequent deletions/duplications at the place of residence, and thus, a patient sample enriched with point pathogenic variant and depleted with frequent deletions is included. A post-hoc analysis of patients from different regions diagnosed in different years will show whether the mutational spectrum in Russia is the result of a biased sample or not.
As the current manuscript only represents the preliminary data analysis, the registry will be expanded in the future, thus, it may help examine incidence, prevalence, mortality and survival rate in patients with DMD in Russia as a whole and in separate Russian regions; tracking the routing of patients from diagnosis to disease outcome; assessing the delay in diagnosis; detection of diagnostic delay in Russia, the average age of symptoms onset, age of DMD diagnosis and treatment start, % misdiagnosis on different steps; assessing long-term outcomes following different treatment approaches for DMD; monitoring effectiveness and appropriateness (according to International Standards of Care) of prescribed diagnostic methods and treatment funded by public budget; determination of prevalence of different types of DMD pathogenic variants in Russia. An additional limitation is the incomplete data collection concerning corticosteroid use along with other treatment options and their outcome, and data on loss of ambulation, which is the subject of future research in the Registry.
Thus, in this study, we present the first results of the work of the National DMD/MDD Registry in Russia. The analysis revealed the specifics of the mutational spectrum in the Russian population and a large proportion of patients who can be prescribed the targeted therapy. The second stage of the Registry data analysis will also include dynamic observation collected by an expert neurologist and clinical geneticist at the referral site. Further work of the Registry will be aimed at increasing the number of patients both retrospectively and prospectively and at the standardized collection of clinical data of patients, both at the time of diagnostics and over time.
Highlights
There are currently no reliable epidemiological data on Duchenne/Becker Muscular Dystrophy in the Russian Federation.
This study presents the first results of the National Registry of Patients with Duchenne/Becker Muscular Dystrophy in Russia.
Almost half of the patients in the Registry live in the Central and Volga Federal Districts, which account for 25.1% and 24.3%, respectively.
Among the 626 patients in the Registry, the most common cause of the disease (52.4%) was large deletions of one or more exons.
The delay in diagnosis (time from the onset to the confirmed diagnosis) in Russia was 24.3 months.
Supplemental Material
sj-docx-1-jnd-10.1177_22143602251356189 - Supplemental material for Molecular and genetic characteristics of patients from the National Registry of Duchenne/Becker Muscular Dystrophy in the Russian Federation: Pilot analysis
Supplemental material, sj-docx-1-jnd-10.1177_22143602251356189 for Molecular and genetic characteristics of patients from the National Registry of Duchenne/Becker Muscular Dystrophy in the Russian Federation: Pilot analysis by Peter A Sparber, Elena V Zinina, Olga Shchagina, Aleksander V Polyakov and Sergey I Kutsev in Journal of Neuromuscular Diseases
Supplemental Material
sj-docx-2-jnd-10.1177_22143602251356189 - Supplemental material for Molecular and genetic characteristics of patients from the National Registry of Duchenne/Becker Muscular Dystrophy in the Russian Federation: Pilot analysis
Supplemental material, sj-docx-2-jnd-10.1177_22143602251356189 for Molecular and genetic characteristics of patients from the National Registry of Duchenne/Becker Muscular Dystrophy in the Russian Federation: Pilot analysis by Peter A Sparber, Elena V Zinina, Olga Shchagina, Aleksander V Polyakov and Sergey I Kutsev in Journal of Neuromuscular Diseases
Footnotes
Acknowledgements
Technical editing of the manuscript and preparation for submission were done with support from the Medical Adviser's Group.
Authors’ contribution
Study conception and design, data collection, analysis and interpretation of results, and draft manuscript preparation were done equally. All authors reviewed and approved the final version of the manuscript.
Funding
The author disclosed receipt of the following financial support for the research, authorship and/or publication of this article: The National Registry of patients with DMD/BMD in Russia was initiated as an IIT “Observational prospective and retrospective study: Duchenne Muscular Dystrophy Registry (Russian Federation)” and was sponsored and funded by PTC Therapeutics LLC. The research was carried out within the state assignment of the Ministry of Science and Higher Education of the Russian Federation for RCMG.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
The data supporting the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.