
Abstract
Background:
Eteplirsen, the first FDA-approved RNA-modifying therapy for DMD, is applicable to ∼13% of patients with DMD. Because multiple exonic deletions are amenable to exon 51 skipping, the isoforms resulting from the various exon 51-skipped transcripts may vary in stability, function, and phenotype.
Objective/Methods:
We conducted a detailed review of dystrophinopathy published literature and unpublished databases to compile phenotypic features of patients with exon 51 “skip-equivalent” deletions.
Results:
Theoretically, 48 different in-frame transcripts may result from exon 51 skipping. We found sufficient clinical information on 135 patients carrying mutations that would result in production of 11 (23%) of these transcripts, suggesting the remainder have not been identified in vivo. The majority had mild phenotypes: BMD (n = 81) or isolated dilated cardiomyopathy (n = 3). Particularly interesting are the asymptomatic (n = 10) or isolated hyperCKemia (n = 20) patients with deletions of exons 45– 51, 48– 51, 49– 51 and 50– 51. Finally, 16 (12%) had more severe phenotypes described as intermediate (n = 2) or DMD (n = 14), and 6 reports had no definitive phenotype.
Conclusions:
This review shows that the majority of exon 51 “skip-equivalent” deletions result in milder (BMD) phenotypes and supports that exon 51 skipping therapy could provide clinical benefit, although we acknowledge that other factors, such as age at treatment initiation or ongoing standard of care, may influence the degree of benefit.
INTRODUCTION
Duchenne muscular dystrophy (DMD) has an incidence of approximately 1 : 5000 live male births, and is an X-linked, slowly progressive, fatal neuromuscular disease caused by mutations in the DMD gene [1, 2]. Over 5,000 DMD mutations are known, with large exon deletions and duplications accounting for ∼80% of these mutations and small point mutations accounting for ∼20% [3, 4]. These mutations cause the dystrophinopathies including Duchenne muscular dystrophy (DMD), intermediate muscular dystrophy (IMD), Becker muscular dystrophy (BMD), and – more rarely – isolated hyperCKemia and isolated cardiomyopathy. DMD is clinically defined as a boy who loses ambulation prior to 13 years whereas BMD refers to a boy who loses ambulation after 16 years, though these definitions have some variability [5, 6]. The major determinant of phenotype is the reading frame rule, which states that mutations resulting in an mRNA with a disrupted reading frame (“out-of-frame”) result in no functional protein and a more severe DMD phenotype. Conversely, “in-frame” mutations typically allow for production of an internally shortened but functional protein and are predicted to cause a milder BMD phenotype. This rule is accurately predictive in ∼90% of DMD patients, although it is less consistently predictive of BMD (with values of 56– 91% in different cohorts) [4, 7].
Several current therapeutic agents aim to take advantage of this rule by restoring the reading frame to allow for production of an internally shortened but likely functional dystrophin protein. One such FDA approved agent is eteplirsen (EXONDYS 51). Eteplirsen is a phosphorodiamidate morpholino oligomer (PMO) antisense therapeutic that skips exon 51 to restore the reading frame in ∼13% of patients with DMD who have mutations amenable to exon 51 skipping [8]. In this subset of DMD patients treated with eteplirsen, studies have shown increased dystrophin expression and a slowing of disease progression on ambulatory and respiratory measures in comparison to historical controls [8–11].
The reading frame rule is not always an accurate predictor of phenotype, as shown by patients with in-frame deletions of exons 45 to 47 (Δ45– 47) or Δ45– 48 who have milder BMD phenotypes, versus patients with in-frame Δ45– 46 deletions who have severe DMD phenotypes [12]. This clinical evidence highlights the potential variability in protein function after treatment in specific exon skippable mutations as well as the inability to extrapolate patient outcomes solely based on the reading frame rule [12]. We thus sought evidence from published literature and unpublished databases to obtain clinical data from exon 51 “skip-equivalent” patients in order to determine the best estimate of clinical outcome for each skip-amenable mutation.
MATERIALS AND METHODS
Editorial policies and ethical considerations & data sharing
For each database included, each database investigator attested to approval from the relevant institutional ethics board. Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Literature search and inclusion/exclusion criteria
On March 28th, 2017 and again on May 30th, 2019 (as an update), a literature search of PubMed was conducted using the following query: dystroph*[TIAB] AND (Duchenne[TIAB] OR Becker*[TIAB] OR Intermediate[TIAB]) AND (mutation[TIAB] OR MLPA[TIAB] OR ligation[TIAB] OR CGH[TIAB] OR “comparative genomic hybridization” [TIAB]). These search terms were discussed and decided upon by the authors to provide the most extensive list of articles that would meet our minimal criteria (boys with a dystrophinopathy in whom genetic testing had been completed) for in depth evaluation. Titles and abstracts of the identified articles were reviewed to ensure that only relevant publications were included. Articles were excluded if genetic testing for dystrophinopathy was restricted to prenatal screening and no clinical postnatal data were reported, or if the articles only discussed female carriers. In addition, studies that focused on other muscular dystrophies or non-human DMD models were excluded. A total of 196 articles were identified and were reviewed to identify patients with genetically confirmed exon 51 “skip-equivalent” mutations in the DMD gene.
Clinical database and registry search and inclusion/exclusion criteria
As a second source of clinical information, we queried several large dystrophinopathy databases and registries for relevant cases, only after approval from the relevant institutional ethics board, as noted above. Of note, numerous clinical databases were invited and those who chose to participate were the following: the French UMD-DMD-France database [13], the United Kingdom North Star database, Duchenne Parent Project Italy (DMD Diagnostics Project/UNIFE), the Duchenne Registry (previously Duchenne Connect), and the United Dystrophinopathy Project (UDP)/University of Utah database. To protect patient privacy, generic identifiers were assigned to each patient with a genetically confirmed exon 51 “skip-equivalent” mutation in the DMD gene whose data was extracted from each database. Female carriers were excluded from the analysis. To avoid duplication, if patient data were published in an article identified during the literature search, or were present in another database, these data were not re-entered into the clinical data sheet. There was no duplication of any patient either between multiple databases, publications or between databases and publications.
Clinical data coding, information capture and patient classification
Full-text versions of the relevant articles were allocated for review by a subset of authors or contributors (MAW, JPD, RBY, KL, EOR). Individual or groups of curators reviewed data from respective databases as follows: UDP/Utah, RBW; UMD-DMD, STG, FL, RBY; UK North Star, JPD; UNIFE, AF; and Duchenne Registry, AM. To assess duplicate records in multiple databases, one investigator (MAW) compared partially de-identified information (date of birth, mutation, and initials if necessary), assuming any records with these same items represented the same mutation patient.
For each article or database patient that was included in our dataset, it was assumed that the diagnosis of DMD, IMD, BMD, isolated hyperCKemia, isolated cardiomyopathy or asymptomatic was correct. In cases for which a clear discrepancy existed between the diagnosis and the clinical information provided in the article/database, the dystrophinopathy was reclassified based on the age at loss of ambulation. Patients who lost ambulation at age <13 years were classified as having DMD, patients who lost ambulation at age ≥13 years but <16 years were classified as having IMD, and patients who lost ambulation at age ≥16 years were classified as having BMD. Patients with only a family history, no symptoms, and an unknown CK were classified as asymptomatic. Patients with only an elevated CK or an elevated CK with minimal/mild cramping were classified as hyperCKemia. Patients with dilated cardiomyopathy with or without elevated CK and with or without minimal/mild cramping (rare episodes of mild muscle discomfort) and no other skeletal muscle symptoms were classified as dilated cardiomyopathy. Patients with more than mild complaints of cramping and/or any other skeletal muscle symptoms were classified as BMD. Some patients who had not lost ambulation and were younger than age 16 at the time of the last follow-up were classified as pending and others were classified as BMD/IMD/DMD based on clinician judgment (utilizing the clinician’s knowledge of the family history and the patient’s functional data). If a patient diagnosis was deemed ambiguous, the patient would have been flagged for a consensus discussion with a working group consisting of a subset of the authors (same authors for every review). If insufficient data were available to clearly reclassify the phenotype, the default course of action was to defer to the diagnosis reported by the article’s authors or database entry. Due to the lack of cardiac, nocturnal ventilation and cognitive and behavior information available, the provided diagnosis was treated as correct – there was no reclassification of these data.
A clinical data sheet was created as a central repository to house the genotypic and phenotypic data extracted from the published literature search and clinical databases. The rationale for using the clinical data sheet was to improve efficiency of data entry and ease of data retrieval compared with compilation of case reports. The clinical data sheet also allowed the identification of mutations for which no data were reported in the literature or in clinical databases, mutations reported only in the literature or only in a clinical database, or mutations listed in both.
The authors agreed by consensus that the following fields would be used to populate the clinical data sheet: exon 51 “skip equivalent” deletion (eg, exon 3 to 51 deletion); reference (eg, PubMed identifier or name of clinical database); patient identifier as used in the article or patient identification number used in the clinical database; whether all exons were interrogated (yes/no/unknown); method to detect the mutation (MLPA, CGH, RT-PCR, or other); clinician diagnosis (DMD, BMD, IMD, etc); ages at diagnosis, report/last follow up, and loss of ambulation; cardiomyopathy (history, age at diagnosis, last left ventricular ejection fraction, last shortening fraction); nocturnal ventilation (history, age at initiation); biopsy (history, result of dystrophin immunofluorescence or immunohistochemistry, result of dystrophin Western blot); cognitive impairment and author comments. Each record entered into the clinical data sheet contained information on the mutation and at least one clinical data field. A formal risk of bias assessment was not completed because the articles included were not randomized studies, but were case reports and descriptive reviews.
RESULTS
There are theoretically 48 potential in-frame rearrangements of the DMD gene that represent exon 51 “skip-equivalent” deletions (Table 1). In total, clinical information was available for 136 patients representing 11 (23%) of these predicted in-frame deletions. Literature review revealed 6 “skip-equivalent” deletions with 46 patients identified, and database review revealed 11 “skip-equivalent” deletions with 90 patients identified [14–21]. Of the 136 total patients, a total of 22 patients required a diagnosis reclassification (following the definitions/criteria discussed in Methods, above), including 16 from the literature search and 6 from the database search. All diagnosis reclassifications were to a milder dystrophinopathy phenotype. These included 11 “BMD” reclassified as “hyperCKemia”, 6 “BMD” reclassified as “asymptomatic”, and 5 “asymptomatic” reclassified as “hyperCKemia”.
List of all theoretical 51 skip-equivalents. Bold and underlined cells equate deletions found in this systematic review
As expected, the majority of the 136 patients (84%) had milder phenotypes such as BMD (n = 81), hyperCKemia (n = 20), isolated dilated cardiomyopathy (n = 3), or asymptomatic (n = 10) (Table 2, Fig. 1). A total of 16 patients had a more severe phenotype of DMD (n = 14) or IMD (n = 2). Of the remaining 6 patients, all were from databases (UDP, n = 5 and UMD-DMD-France, n = 1) and all were still ambulant and of too young of an age to assign a definitive diagnosis. (Table 2).
Core clinical data on 51 skip-equivalents
Abbreviations include: Dx = diagnosis, Y = yes, N = No, NR = Not reported, LVEF = left ventricular ejection fraction, SF = shortening fraction, Nl = normal, ADHD = attention deficit hyperactivity disorder, ADD = attention deficit disorder, WCPT = wheelchair part time, DD = developmental delay, LD = learning disability, ID = intellectual disability. In cells that are blank, this represents data that was not reported. *denotes member of family mentioned in the text; §listed in source document as a range.
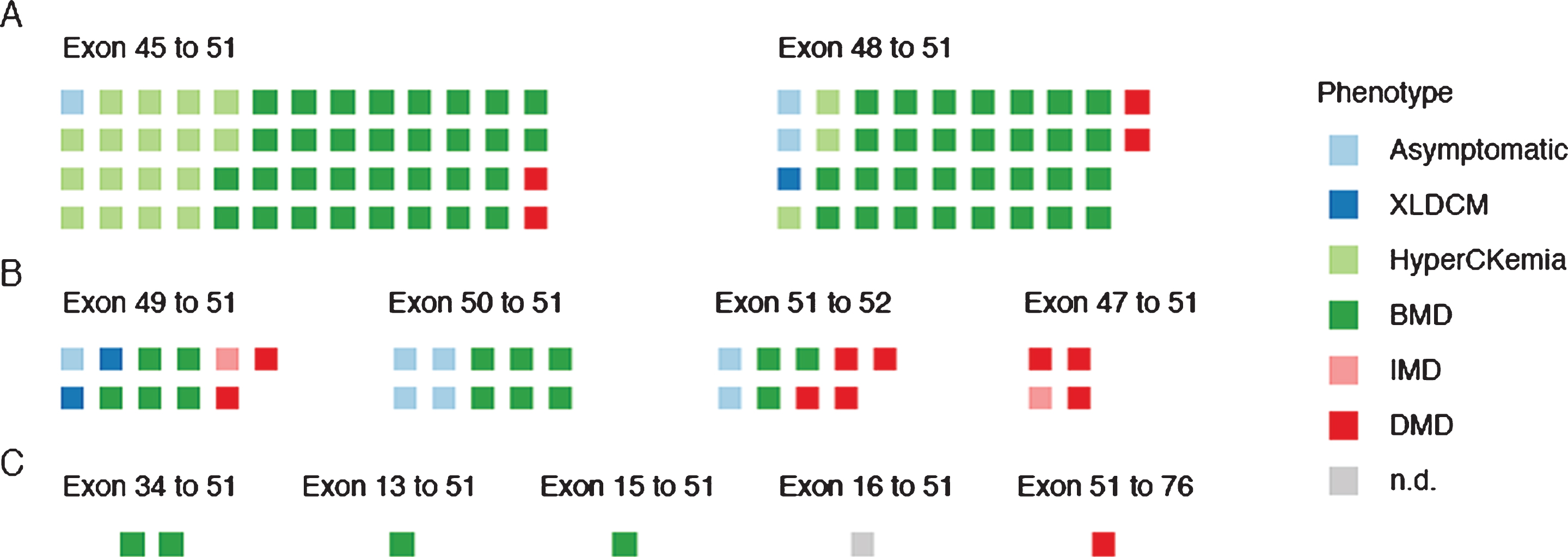
Distribution of phenotypes for each deletion subset with at least four patients. A) Individual patients are grouped by Δ45– 51 (n = 52) or Δ48– 51 (n = 38) skip-equivalent deletions and their phenotypes are represented by colored squares. Patients with pending clinical phenotypes were not included. B) Patients grouped by Δ49– 51 (n = 11), Δ50– 51 (n = 10), Δ51– 52 (n = 9), or Δ47– 51 (n = 4) skip-equivalent deletions. C) Patients grouped by Δ34– 51 (n = 2), Δ13– 51 (n = 1), Δ15– 51 (n = 1), Δ16– 51 (n = 1), or Δ51– 76 (n = 1) skip-equivalent deletions. Additional information on the extent of genetic analysis for each patient can be found in the supplemental table.
The “skip-equivalent” deletions with the largest number of reported patients were Δ45– 51 (n = 56) and Δ48– 51 (n = 39). Both had patients with almost exclusively mild phenotypes, with no IMD and only 2 DMDs present in each. Family structures could not be determined in many reports, but phenotypic variability was reported within one family with Δ48– 51 deletions, in which a grandfather in his 80’s was reported as entirely asymptomatic, one grandson had an isolated cardiomyopathy diagnosed at age 23, and another grandson had skeletal muscle weakness, elevated CK of 3,000, and an attention deficit hyperactivity disorder diagnosis at age 9.
The next most represented “skip-equivalent” deletions, with at least 4 patients reported, were Δ47– 51, Δ49– 51, Δ50– 51 and Δ51– 52. All reported patients with a Δ50– 51 deletion had mild phenotypes (BMD = 6/10, asymptomatic = 4/10), but more variability was seen with the other deletions. Of nine patients found with deletions of Δ51– 52, two were classified as asymptomatic, three were classified as BMD while the remaining four were classified as DMD. The largest variability in phenotype was seen in Δ49– 51 deletions, with all possible phenotypes reported except hyperCKemia. The reported deletion most uniformly associated with a more severe phenotype was Δ47– 51; among 4 reported patients, 3 were described as DMD and 1 as IMD, but only 1 patient had lost ambulation at the time of report. Although only represented by one or two patients, Δ13– 51 and Δ34– 51 deletions were exclusively BMD and the Δ51– 76 deletion was DMD. Only one patient was found with either a Δ15– 51 or Δ16– 51 deletion and insufficient clinical data prevented definitive classification for the patient with the Δ16– 51 deletion, however, he was still walking at age 12.6 years, suggesting a classification of at least IMD.
A reported DMD deletion and a clinical description or classification were required for inclusion in this review. Other clinical information, including the method of mutation analysis, varied significantly (supplemental data), and exon deletions Δ16– 51 (n = 1) and Δ34– 51 (n = 2) had no further clinical information other than the minimum required for inclusion in this review. The method of detection was not provided in 14 reports (10%), and only 65% (n = 88) had what we consider to be complete exon copy number analysis, in which all exons are interrogated. The remaining patients had genetic diagnosis via multiplex PCR with borders defined, but not all exons interrogated (n = 22) or without defining borders (n = 11). The implications of incomplete analysis are of particular importance in considering cases categorized as DMD. Among the 14 patients categorized as DMD, 10 patients had complete exon copy number analysis. The remaining 4 patients either had multiplex PCR with borders defined (Δ47– 51 & Δ51– 76) CGH (Δ49– 51), or multiplex PCR without borders defined (Δ51– 52).
Cardiomyopathy is a key phenotypic component of the dystrophinopathies and information on the presence or absence of cardiomyopathy was available in 54% (n = 74) of the reported patients. A definite cardiomyopathy was reported in 14% (n = 10) and was diagnosed at an average age of 34 years, although this is a limited report that spans several deletions: Δ45– 51, Δ47– 51, Δ48– 51, Δ49– 51 and Δ51– 52. Of the 61 patients who did not have a diagnosis of cardiomyopathy, only 16% (n = 12) were over age 20, so it is unknown if they may develop cardiomyopathy later in life, although development of cardiomyopathy after age 20 would still be considered a milder phenotype. The highest proportion of older patients without cardiomyopathy was seen in Δ45– 51, Δ48– 51 and Δ49– 51 deletions.
Dystrophinopathies also impact respiratory function, and though this was reported less frequently than cardiac status, information on nocturnal ventilation status was available in 45% (n = 61) of the reported patients. Use of nocturnal ventilation was surprisingly low at 5% (n = 3) with ages at initiation of 38 years (Δ13– 51), 14 years (Δ45– 51) and 54 years (Δ49– 51). Information on pulmonary function testing was not available. This low number could reflect a milder phenotype or may not have been reliably reported.
Information on cognition and behavior was available in 38% (n = 51) of the reported patients with cognitive involvement noted in 23% (n = 12). No information on cognition was available for Δ15– 51, Δ16– 51, Δ34– 51, or Δ51– 52 deletions. Of those deletions with some cognitive data reported, no cognitive involvement was reported for Δ13– 51 or Δ47– 51, though the sample size was small, 1 of 1 and 2 of 5, respectively. Cognitive involvement was reported in patients with deletions of Δ45– 51, Δ48– 51, Δ49– 51, Δ50– 51 and Δ51– 76 and included attention deficit disorder/attention deficit hyperactivity disorder, speech and language difficulties, intellectual deficiency, autism, or learning disability (see supplemental data). This presumably represents the concomitant deletion of the translational initiation site of the predominant brain isoform Dp140, located in exon 51, as well as deletion of the Dp116 and Dp71 promoters in the case of the Δ51– 76 patient. Although this data set is limited, it does seem to suggest that the incidence of cognitive and behavioral issues may be lower in those with exon 51 “skip-equivalent” deletions.
Muscle biopsy data
Information on dystrophin expression was limited. Whether or not a muscle biopsy was performed was reported in 57% of the patients (n = 78), half of whom (n = 39) had a muscle biopsy as part of their evaluation (supplemental data). In general, the muscle biopsy reports correlated with clinical phenotype as expected by revealing the presence of variable amounts of dystrophin, although detailed correlation was impossible due to variability in reporting, and variable or undescribed assay methods. For example, of the three patients listed as asymptomatic who had a biopsy, two had dystrophin noted to be present either in high quantity or of normal size, whereas the third had no comments.
Frequency of skip-amenable mutations
As an additional analysis, we determined the number of patients present in these databases with out-of-frame mutations amenable to exon 51 skipping therapy, although the query did include older and deceased patients. A total of 755 patients with exon 51 skip-amenable mutations were identified collectively (Table 3). As expected, exon Δ45– 50 (29%), Δ48– 50 (25%) and Δ49– 50 (20%) were the most represented [3].
Database Counts of skip-amenable mutations
This table represents the number of skip-amenable mutations present in each of the databases queried for this report.
DISCUSSION
The first FDA approval of a RNA-modifying therapy for DMD is an exciting development and is applicable to ∼13% of the DMD community with “51 skip-amenable” whole exon deletions [8]. While not yet evaluated clinically, exon 51 duplications and some isolated point mutations found in exon 50 or 52 splice sites may also benefit from exon 51 skipping therapy. Furthermore, development of antisense oligonucleotides (ASOs) to skip additional exons could provide treatment options for up to ∼80% of patients with DMD [22] and clinical trials are currently underway to evaluate exon 51, 45 and exon 53 skipping ASOs using a variety of backbone chemistries (NCT02500381, NCT03532542, NCT02310906, NCT02530905, NCT03167255, NCT03680365, NCT02667483). While there are 48 whole exon mutations theoretically amenable to exon 51 skipping (Table 1), only eleven of these skip amenable mutations were identified in patients in the searched databases, with the most commonly reported mutations being Δ45– 50, Δ48– 50, Δ49– 50, Δ50 and Δ52 (Table 3). These findings are consistent with frequencies reported previously, further supporting that the majority of patients amenable to exon 51 skipping have one of these 5 mutations [3, 22].
Our report focuses on the phenotypes associated with mutations equivalent to those that would result from therapeutic exon skipping. Utilizing literature reports as well as patient database resources to search for in-frame, exon 51 skip-equivalent patients, this fairly comprehensive report only identified clinical information on 23% (11 out of 48) of the skip-equivalent deletions. This absence of data may be because the 37 remaining mutations are exceedingly rare or that the resulting clinical phenotypes are so mild that the mutations remain unreported.
The available clinical data on patients with these 51-skip equivalent mutations shows that the majority have mild phenotypes (with the majority over age 14 and still walking at the time of last report with a max age of 80.7). A subset of patients (12%) had severe phenotypes despite their predicted “in-frame” mutation. This highlights the complexity of the dystrophinopathies and a need for nuanced understanding of the “reading-frame” rule. When this rule is used clinically, it is predictive ∼90% of the time in DMD, and 56% to 91% of the time in BMD [3, 13]. The wider range reported as a predictive value for BMD may reflect different biases of categorization among the cohorts studied, with higher values in cohorts defined from mutational databases with limited clinical data [3, 13], and a lower predictive value reported in a large cohort in which categorization was made primarily on clinical severity [7]. Regardless, it is clear that although certain mutations detected by standard clinical analysis may be predicted to have the effect of truncating the reading frame, the ultimate phenotype may be influenced by a variety of factors, including the inherent “leakiness” of splicing seen in some exons [23] the effect of the predicted frame-terminating mutations on splicing signals [24], and the stability and tridimensional structure of the resultant protein [12, 25], with the latter associated with variability in size, abundance, and localization of in-frame BMD-associated dystrophins that has been demonstrated in human biopsy samples [26, 27].
There are several limitations to our dataset. One is that clinical information was limited in several of the patients identified in the literature with some patients given a diagnosis of DMD simply if they were younger than age 12 and had not yet lost ambulation. A second is that one of the databases (the UK North Star Database) only enrolled patients with a diagnosis of DMD, potentially skewing the results toward a more severe phenotype. Perhaps the greatest limitation to our dataset is the fact that 35% of the included patients did not have genetic testing that interrogated all exons, leaving the possibility that an additional exon could have been deleted or that the patients may have harbored a second mutation. It is known that in rare cases of analysis using MLPA or PCR-based methods, allele drop out may occur due to exonic SNPs causing failure of amplification and consequently erroneous deletion definition [28]. As a result, most labs using MLPA or similar approaches perform a PCR using a different set of primers to confirm the absence of exons that define deletion limits [7]. The majority of data that we reviewed was obtained prior to publication of the care standards for Duchenne muscular dystrophy, which propose interrogation of all exons, and confirmation of exon limits [28, 29]. As a result, we expect that in the future, inaccurate genetic diagnosis will be an increasingly rare issue for patients. Had this been a prospective study, independent validation via RNASeq/RT-PCR could have been conducted on all samples to confirm mRNA structure and exclude a second point mutation. However, it is important to note that when multiple patients with the same genotype have been reported with very similar phenotypes, numerous secondary mutations are unlikely. We also want to highlight that of the 10 asymptomatic patients, 9 had a definitive genetic diagnosis (2 via MLPA and 7 via multiplex PCR with borders defined). We also note that the data for cardiomyopathy, nocturnal ventilation and cognition and behavioral was somewhat limited and variable between patients. Thus, these data should be interpreted accordingly.
An explanation for the relatively high numbers of patients with reported DMD phenotypes (n = 9) for the Δ47– 51, Δ49– 51, Δ51– 52 deletions is unclear. The most notable of these cohorts is the exon Δ47– 51 deletion group (n = 4), in which all ascertained patients had more severe phenotypes (three with DMD, and one with IMD), suggesting that this deletion may result in a less stable or less functional protein. Two patients had genetic analysis via MLPA, one patient via multiplex PCR with borders defined, and one patient via an unreported method of genetic testing. Thus in one patient, we do not know if genetic analysis has thoroughly defined the deletion and in the other three patients, a second mutation or non-contiguous rearrangement— although exceedingly rare— have not been excluded. Unfortunately, given the retrospective nature of this report, skeletal muscle tissue analysis was not possible. Given these limitations, despite the findings of a severe phenotype in these reported patients it is premature to draw any conclusions about the use of exon 51 skipping therapy for patients with exon Δ47– 50 deletions, and confirmation awaits further assessment in clinical practice.
Although this study aimed to correlate phenotype with specific DMD deletions, we note that phenotype may also be influenced by genetic modifiers. Genome wide association studies on large dystrophinopathy datasets have identified a few modifiers of age at loss of ambulation, hand grip strength, and responsiveness to glucocorticoids. The IAAM haplotype of the LTBP4 genotype that is associated with an ∼2 year delay in time to loss of ambulation in this population was first reported in the United Dystrophinopathy dataset and has been confirmed in other dystrophinopathy datasets [30–33] and TCTEX1D1 has just recently been reported as a modifier of ambulation [34]. The SPP1 TT genotype has been associated with a more robust response to glucocorticoid therapy but this has not yet confirmed in additional datasets, nor has the claim that it may also contribute to longer preservation of hand grip strength [32, 33]. Newer evidence suggests that THBS1 may also play a role in mitigating phenotype and further work to identify other potential modifiers is underway [35]. Although genetic modifiers may have influenced the reported phenotypes in our data set, particularly in outliers, genotyping of known modifiers was not possible in this retrospective analysis.
A further potential source of variability in our analysis is differences in standard of care provided to these patients from diverse regions. Time to diagnosis, age at initiation of glucocorticoids when initiated, length of glucocorticoid use, and management of respiratory and cardiac function all have significant impact on functional outcomes and are known to vary between sites [36–40]. Care guidelines were established in 2010 and updated in 2018, yet not all providers are aware of or are following these guidelines and an optimal glucocorticoid dosing regimen is still being determined (FOR-DMD NCT01603407) [6, 41– 43]. Thus, management of each patient reported here is another variable influencing phenotype in these patients.
Overall, this review largely highlights what has been previously reported in the literature, supporting our understanding of the structure-function relationship of the dystrophin protein [44]. Specifically, the absence of large portions of the spectrin repeat-containing central rod domain may have limited impact on skeletal muscle pathology. For example, the report of a patient missing exons 13 to 51 with features of BMD – walking into his late 20’s – indicates that even very large deletions sparing the amino-terminal actin binding domain 1 can result in a relatively mild phenotype. In addition, the large number of patients with a 45 to 51 deletion reported as BMD (in two instances with very late ages of diagnosis: 67 and 78 years old) indicates that patients missing the canonical nNOS binding domain, which is coded for by exons 42– 45, can experience a mild phenotype. On the other hand, certain protein domains are critical to dystrophin function. For example, our finding that a patient with a deletion of exons 51 to 76 was diagnosed as DMD and stopped walking before age 10 years provides additional evidence of the importance of the dystroglycan binding domain, supporting the evidence for the criticality of a functional cysteine-rich beta-dystroglycan binding domain as previously demonstrated in both DMD patients [45, 46] and in the mdx mouse model [47, 48]
The phenotypic variability in patients with “skip-equivalent” mutations reported here suggests that treatment response to exon 51 skipping may also be variable and impacted by multiple factors in addition to DMD genotype. Phenotypic variability seen in patients with the same exon deletions supports the need for continued evaluation of genetic modifiers and internationally accepted standards of care for this heterogeneous population. Nevertheless, the repeated observation of patients with a BMD phenotype in association with skip-equivalent mutations— particularly those more common mutations that are well-represented in existing databases— supports that exon 51 skipping therapy has the potential to provide clinical benefit, although we acknowledge that other factors, such as age at treatment initiation (since the BMD “skip-equivalent” patients produce dystrophin from birth) or ongoing standard of care (including corticosteroid use), may influence the degree of benefit. We believe that patient-derived information such as that represented here can serve as a resource for pediatricians and neuromuscular clinicians by providing insight into the phenotypic range of patients reported with exon 51 skip-equivalent mutations.
FILNEMUS NETWORK
The French health Finemus network for neuromuscular diseases brings together clinical neuromuscular units and laboratories of Molecular Genetics (see http://www.filnemus.fr/). Members of Filnemus more specifically involved in this study as clinicians (PACA-Réunion-Rhône Alpes Reference Center: F. Audic, B. Chabrol, V. Manel, P. Petiot, E. Salort-Campana, C. Vial; Nord/Est/Ile de France Reference center: B. Eymard, C.Barnerias, A. Behin, F. Chapon, J.M. Cuisset, I. Desguerre, P. Laforet, V. Laugel, AG. Lemoing, P. Sabouraud, C. Vincent-Delorme, JA. Urtizberea; Atlantique/Occitanie/Caraïbes reference center: MC Arne-Bes, S. Audebert-Bellanger, C. Cances, P. Cintas, M. Fradin, R. Juntas-morales, D. Martin, D. Menard, MC. Minot-Myhie, H. Rauscent, A. Toutain, F. Rivier, U. Walther-Louvier) and as Laboratories of Molecular Genetics: V. Biancalana (Hôpital Civil Strasbourg), M. Cossée (CHU Montpellier), AS. Denommé-Pichon, P. Jonveaux (CHRU Nancy), C. Dubourg (CHU Rennes), P. Malzac (Hôpital Timone Enfants), L. Michel-Calemard (Hospices Civils de Lyon), MP. Moizard (CHRU Tours) and S. Schmitt (CHU Nantes Hôtel Dieu).
POTENTIAL CONFLICTS OF INTEREST
Dr. Waldrop served on one advisory board meeting from Sarepta Therapeutics which manufactures eteplirsen. Karin Lucas and Erin O’Rourke are full time employees and stock holders of Sarepta Therapeutics. Ann Martin’s employer, Parent Project Muscular Dystrophy receives funding from Sarepta Therapeutics for their free genetic testing program. Drs. Ferlini, Muntoni and Flanigan have received personal fees and grant support from Sarepta Therapeutics. Dr. Leturcq has received personal fees from Sarepta Therapeutics.
Footnotes
ACKNOWLEDGMENTS
The authors would like to thank the late Joana Pisco Domingos (JPD) for her contribution of DMD patients from the UK North Star database and AFM-Telethon for supporting the UMD-DMD-France database. The literature search was conducted by Peloton Advantage with funding from Sarepta Therapeutics.