
Abstract
Duchenne muscular dystrophy (DMD) is caused by loss-of-function mutations to the gene encoding dystrophin. Restoring the reading frame of dystrophin by removing internal out-of-frame exons may address symptoms of DMD. Therefore, the principle of exon skipping has been at the center stage in drug development for Duchenne muscular dystrophy (DMD) over the past two decades. Antisense oligonucleotides (AONs) have proven effective in modulating splicing sites for exon skipping, resulting in the FDA approval of several drugs using this technique in recent years. However, due to the temporary nature of AON, researchers are actively exploring genome editing as a potential long-term, single-administration treatment. The advancements in genome-editing technology over the last decade have boosted this transition. While no clinical trials for exon skipping in DMD via genome editing have been conducted as of this writing, preclinical studies show encouraging results. This review describes the preclinical landscape of genome editing for exon skipping in DMD treatment. Along with highlighting the adaptability of genome editing in exon skipping, this review also describes delivery challenges and outlines future research directions that could set a new stage for enhanced therapeutic development in DMD.
Introduction
Duchenne muscular dystrophy (DMD) is among the most prevalent genetic diseases and results in premature death. 1 It is primarily attributed to out-of-frame intragenic deletions or duplications affecting one or multiple exons within the gene encoding dystrophin (DMD gene, denoted as italicized uppercase for human DMD and Dmd for the mouse and canine gene). These out-of-frame mutations stop protein translation prematurely, leading to non-functional dystrophin protein.2,3 The devastating impact of DMD has prompted extensive efforts to explore numerous potential treatment avenues, resulting in significant milestones in DMD medicine research.4–8
Until now, corticosteroid therapy remains the gold standard of care for DMD management, albeit with inherent limitations.6,8 Emerging dissociative steroid treatments, such as vamorolone, offer a promising alternative by mitigating steroid-related adverse effects. 9 Concurrently, DMD research efforts have also focused on restoring the disrupted DMD gene. Therapeutic approaches that aim to restore partially functional muscle dystrophin in patients with DMD focus on one of three approaches: 1. stop codon read-through; 2. micro/mini-dystrophin gene replacement using viral vectors; 3. converting out-of-frame mutations to in-frame mutations (exon skipping; multiple approaches).6,10–12
Among these strategies, exon skipping drugs facilitated by antisense oligonucleotides (AON) have received the most approvals from FDA (Food and Drug Administration) or EMA (European Medicines Agency), mainly due to their safety profile and the unmet need for therapies for DMD. This approach operates on the premise that converting the translational reading frame of the mutated dystrophin protein from out-of-frame to in-frame yields a truncated yet functional dystrophin, addressing the nonfunctional dystrophin characteristic of DMD.13,14 However, the limited applicability of single-exon skipping, constrained by its mutation-specific nature, means that only around 30% of DMD patients may potentially benefit from current therapies, leaving a significant proportion untreated. 15
The most recent FDA-approved treatment involves microdystrophin gene transfer using Adeno Associated Virus (AAV) as a delivery vector. 16 This approach addresses the challenge of mutation specificity inherent in DMD patient populations. 12 While the open-label gene transfer study demonstrated robust microdystrophin expression on muscle biopsy and meaningful functional improvement after one year, 17 long-term sustainability and efficacy of this treatment require further observation and evaluation.
Genome-editing technologies offer great opportunities to cure genetic diseases. The advancement of genome editing, particularly with the emergence of CRISPR technology, has revolutionized biological research and therapeutic possibilities. 18 A recent milestone includes the approval by the FDA of the first CRISPR treatment for sickle cell disease, marking a significant breakthrough in the field of genomic medicine. 19
DMD has been a primary target for gene therapy development, specifically with genome editing-mediated exon skipping showing promise at the preclinical stage.10,20 Given the growing interest and advancements in this area, it is crucial to examine the current state of research on genome editing-mediated exon skipping in DMD. By exploring the latest developments, challenges, and prospects, this review aims to provide insights into the potential of genome editing as a therapeutic strategy for DMD.
Exon skipping as a therapeutic modality
The DMD gene is the largest in the human genome and is characterized by complexity, including multiple transcriptional initiation points (alternative promoters), spanning 79 exons dispersed over 2 megabases of the X chromosome. Over 8585 unique variants have been documented in the DMD gene, with more than 6500 associated with dystrophinopathies. 21 Approximately 60% of mutations leading to DMD occur within mutational hotspots, notably concentrated from exons 45 to 55.15,22
Exon skipping has emerged as a promising therapeutic approach for DMD patients. This strategy aims to restore the disrupted reading frame of DMD transcripts, enabling the synthesis of partially functional, internally deleted Becker-like dystrophin proteins, rather than prematurely truncated non-functional Duchenne dystrophins.13,14 Exon skipping can be accomplished using diverse methods, including exon masking techniques like phosphorodiamidate morpholino oligomers (PMO) AONs or uridine-rich 7 (U7) small nuclear RNA (snRNA) expressing antisense sequences (Figure 1
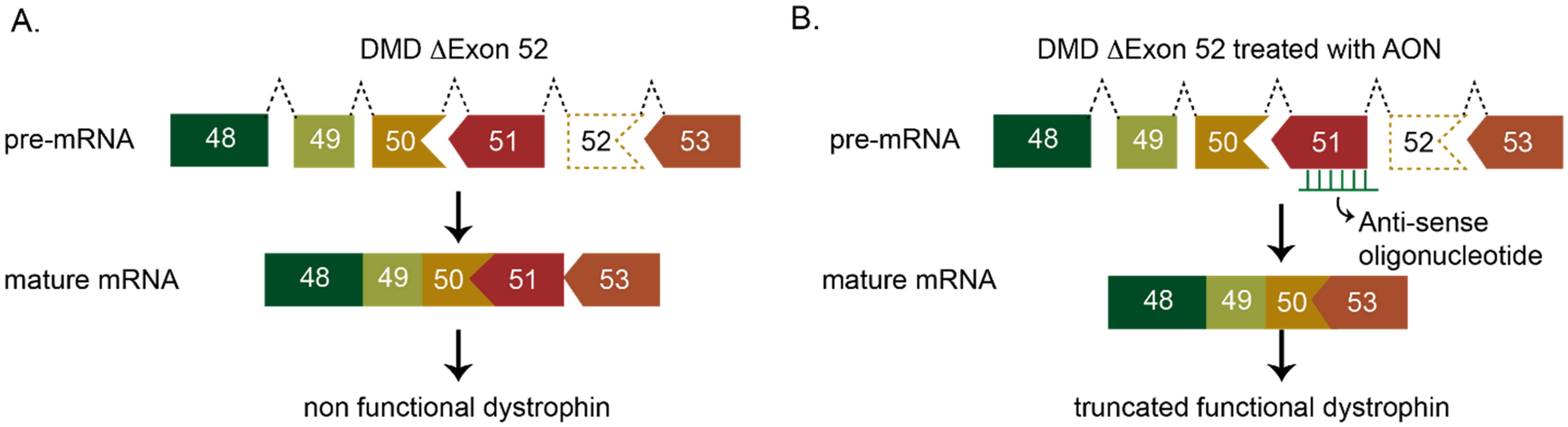
Currently, among the six FDA- or EMA-approved interventional therapies for DMD, four use PMO AON-mediated exon skipping approaches, all of which target exons within the hotspot region spanning exons 45–55 (Figure 2). These therapies include eteplirsen (Exondys 51: NCT02255552, NCT03218995, NCT01540409, NCT04179409), targeting exon 51; golodirsen (Vyondys: NCT04179409); and viltolarsen (Viltepso: NCT04956289, NCT04060199), targeting exon 53; and casimersen (Amondys: NCT04179409), targeting exon 45.24–27 Further studies are underway to confirm and evaluate the long-term efficacy and safety of exon-skipping-mediated therapies (NCT03992430, NCT04687020).
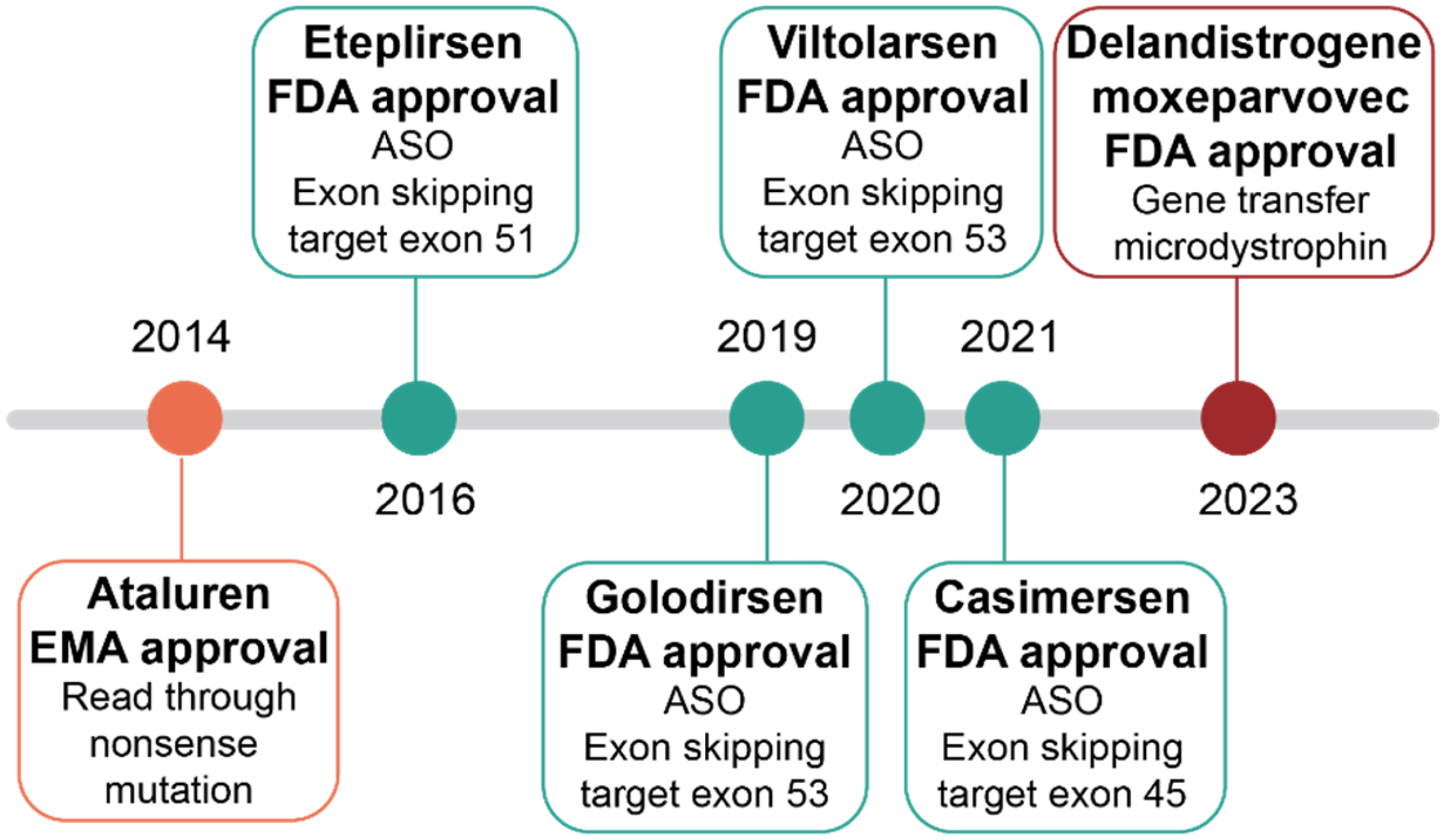
Despite the effectiveness of AON-based exon skipping therapy, its applicability is constrained by its mutation-specific nature. Single-exon skipping treatments can only address a subset of DMD mutations, with each therapy typically applicable to a small percentage of patients, ranging from 8% to 13% of all cases. 15 However, AONs are not curative and only provide temporary effects on RNA requiring multiple administrations. This requires a lifelong healthcare commitment for both patients and hospitals, coupled with notable economic challenges due to the high cost of these drugs.
The recent approval of Delandistrogene moxeparvovec, an AAV gene therapy approach, has changed the landscape of mutation-agnostic options for DMD therapy. Gene replacement therapy does not rely on the principle of exon skipping, however, is based on a shorted dystrophin. Gene replacement therapy for DMD is reviewed extensively elsewhere. 12
Genome editing approach for exon skipping
Genome editing offers a promising avenue for addressing the limitations of AON-based exon skipping therapy by enabling simultaneous targeting of multiple exons and potentially inducing permanent modifications. This capability can lead to sustained therapeutic effects with a single treatment. Moreover, genome editing can potentially address various types of mutations in DMD, such as premature stop codons or exon duplications, thereby broadening its therapeutic scope.
The advancement of programmable nucleases has revolutionized genome editing by providing powerful tools for modifying target genome sequences. These nucleases, including meganuclease (MGN), zinc finger nuclease (ZFN), transcription activator-like effector nuclease (TALEN), and clustered regularly interspaced short palindromic repeats (CRISPR), can be engineered to create double-strand breaks (DSBs) at specific DNA sequences. Subsequently, these breaks are repaired by either non-homologous end joining (NHEJ) or homology-directed repair (HDR). 28
The use of programmable nucleases in DMD research spans more than a decade and has shown potential in restoring DMD gene expression. The advancement of genome editing and various editing methodologies in DMD research has been extensively reviewed elsewhere. 10 While all these nucleases exhibit therapeutic potential, MGN, ZFN, and TALEN demand substantial effort in constructing and testing to engineer a new protein for a single target DNA sequence. In contrast, CRISPR − Cas9 offers advantages by removing protein engineering and design and simply requiring design of a guide RNA (gRNA) for targeting diverse sequences. The adoption of systems like ZFN and TALEN for engineering sequence-specific DNA binding proteins is notoriously more time-consuming compared to the procedures associated with CRISPR − Cas9.11,29 Consequently, CRISPR − Cas9 currently stands as the predominant approach in genome engineering.
Exon skipping in genome editing primarily involves either deleting the mutated exon or disrupting splice sites. The deletion of one or more exons can function similarly to AONs, masking the mutated exon(s) and causing them to be skipped during splicing. In cases of splice site disruptions, indels creation at the splice sites can trigger exon skipping. Ousterout et al. were the first to utilize ZFN to skip exon 51 by deleting the exon, thus restoring DMD gene expression in myoblasts derived from DMD patients with exon 48–50 deletions (Figure 3(A)).
30
In a concurrent study, Li et al. demonstrated the application of TALENs to disrupt the splice acceptor for skipping exon 45 in patient-derived iPSCs (Figure 3(B)
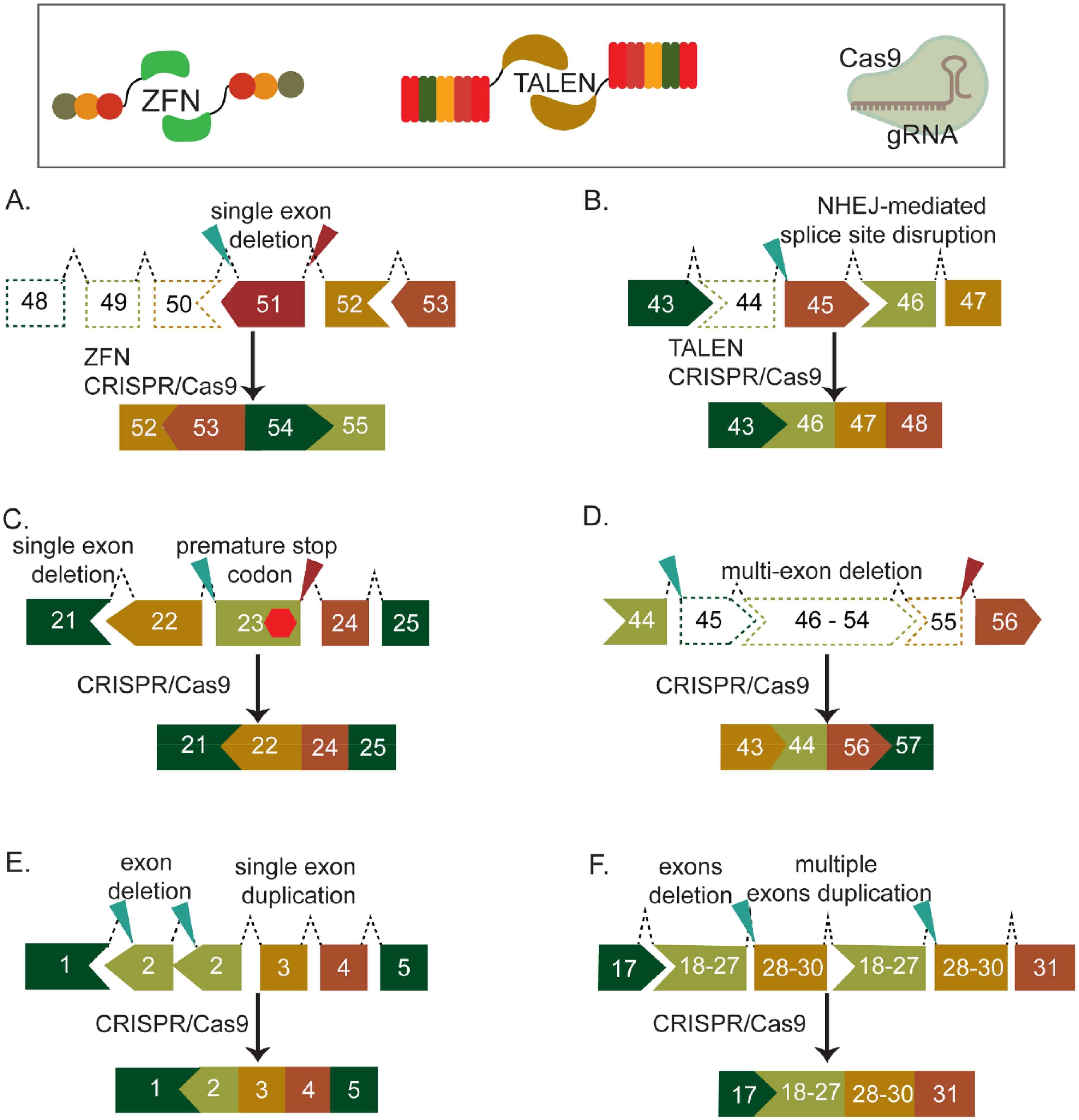
In 2016, three research groups independently reported in vivo delivery of CRISPR − Cas9 for exon skipping, specifically targeting Dmd exon 23 in the mdx mouse model (Figure 3(C)). By using two gRNAs designed specifically for S. pyogenes or S. aureus Cas9, they successfully excised exon 23, which harbored a nonsense mutation. Using AAV as the delivery vector, all groups reported the restoration of dystrophin expression and improvement in muscle function.32–34 After this in vivo proof of concept, an intensified research effort has emerged, demonstrating the potential of CRISPR tools to mediate exon skipping in various animal models of DMD, including canine and pig models.35–37 The in vivo outcomes are promising, ranging from dystrophin restoration to improved skeletal or cardiac muscle function and even prevention of cardiomyopathy.32–36,38–41
CRISPR-Cas9 can also facilitate the induction of multiple exon deletions, mimicking the effect of multi-exon skipping (Figure 3(D)). The first multi-exon deletion was performed using adenovirus as a delivery vehicle targeting exon 21–23, resulting in approximately a 23 kb deletion, successfully restoring Dmd expression in mdx mice. 42 Furthermore, the use of a multiplexed gRNA targeting hotspot mutation in exons 45–55 restored dystrophin expression in vitro in patient-derived myoblasts.43,44 Upon implantation of these myoblasts into mice, sustained expression was observed. 43 Targeting this hotspot is estimated to restore the dystrophin open reading frame in over 60% of DMD patients. 45 The approach of multi-exon deletions for restoring dystrophin expression has also been successful in human iPSC-derived cardiomyocytes by targeting exons 3–9. This has shown normalized Ca2 + transient kinetics. 46 Additionally, in a mouse model carrying a nonsense mutation in exon 53, the deletion of exons 52 and 53 resulted in approximately 34% dystrophin expression in cardiac myofibers. 40 Importantly, this study by Bengtsson et al. compared the feasibility of homology directed repair and exon deletion in vivo.
Building upon Ousterout et al.'s 29 and Li et al.'s previous in vitro validation, 31 exon skipping can also be achieved without resorting to the dual CRISPR-Cas9 method by targeting the splice acceptor site of the exon in vivo. In 2017, Amoasii et al. focused on the splice acceptor site of exon 51 and achieved 60–90% restoration of dystrophin in skeletal or cardiac muscles in ΔEx50 mice and a DMD canine model.37,41 Similarly, in 2019, Min et al. demonstrated exon 45 skipping by inducing indels at the splice acceptor site for exon 45. The systemic delivery of CRISPR-Cas9 using the AAV9 vector was found to restore dystrophin levels to approximately 90% of wild-type levels in TA, triceps, diaphragm, and heart in ΔEx44 mice. 47
A single guide RNA method can also effectively target duplication mutations, facilitating the skipping of all duplicated exons and restoring the reading frame of dystrophin (Figure 3(E) and (F)). This approach has successfully addressed single or multiple exon duplications both in vitro and in vivo. The removal of exon 2 duplication or a duplication spanning exons 18–30 of the DMD gene in DMD patients’ myotubes or Dup18-30 mice resulted in the restoration of full-length dystrophin expression.48,49 The gRNA is designed to attach to two specific spots within the duplication, forming two DSBs. This process leads to the removal of the sequence between the breaks, which covers the entire duplication. Duplication removal could also be accomplished with two gRNAs, each targeting a duplication junction. However, the availability of suitable gRNA target sequences is more restricted in this approach. By using a single gRNA, a broader range of target sequences within the entire duplication sequence becomes accessible, facilitating the design of guides with reduced off-target effects. 49
Despite these promising preclinical findings, certain challenges could hinder the effective use of CRISPR-Cas9 as a therapy. Although Cas9 is directed to create DSBs at specific sites, there is evidence of complex and difficult to measure DSB outcomes, such as unintended large deletions, chromothripsis, chromosomal translocations, and p53 activation.50–56 Advancements in base editing (BE) and prime editing (PE) technologies aim to address some of these challenges by allowing more precise modifications of the DNA sequence without the need for DSBs. Yuan, et al., demonstrated a cytosine base editor (CBE) fused with targeted-AID mediated mutagenesis (TAM) can efficiently convert 90% of invariant guanines to adenines at either 5′ or 3′ splice sites to exclude exon 50 in hiPSCs 57 (Figure 4). The improved TAM-CBE fusion has also shown efficacy in rescuing dystrophic cardiomyopathy in DMD mice, effectively preventing ventricular remodeling. 58 Another application of CBE-TAM confirmed its utility in skipping multiple exons in the DMD gene, with efficiency enhancements achievable through targeting exonic splicing enhancers. 59 Splice site conversion using another BE tool, adenine base editor (ABE), has also been successfully used to induce exon skipping.60–62
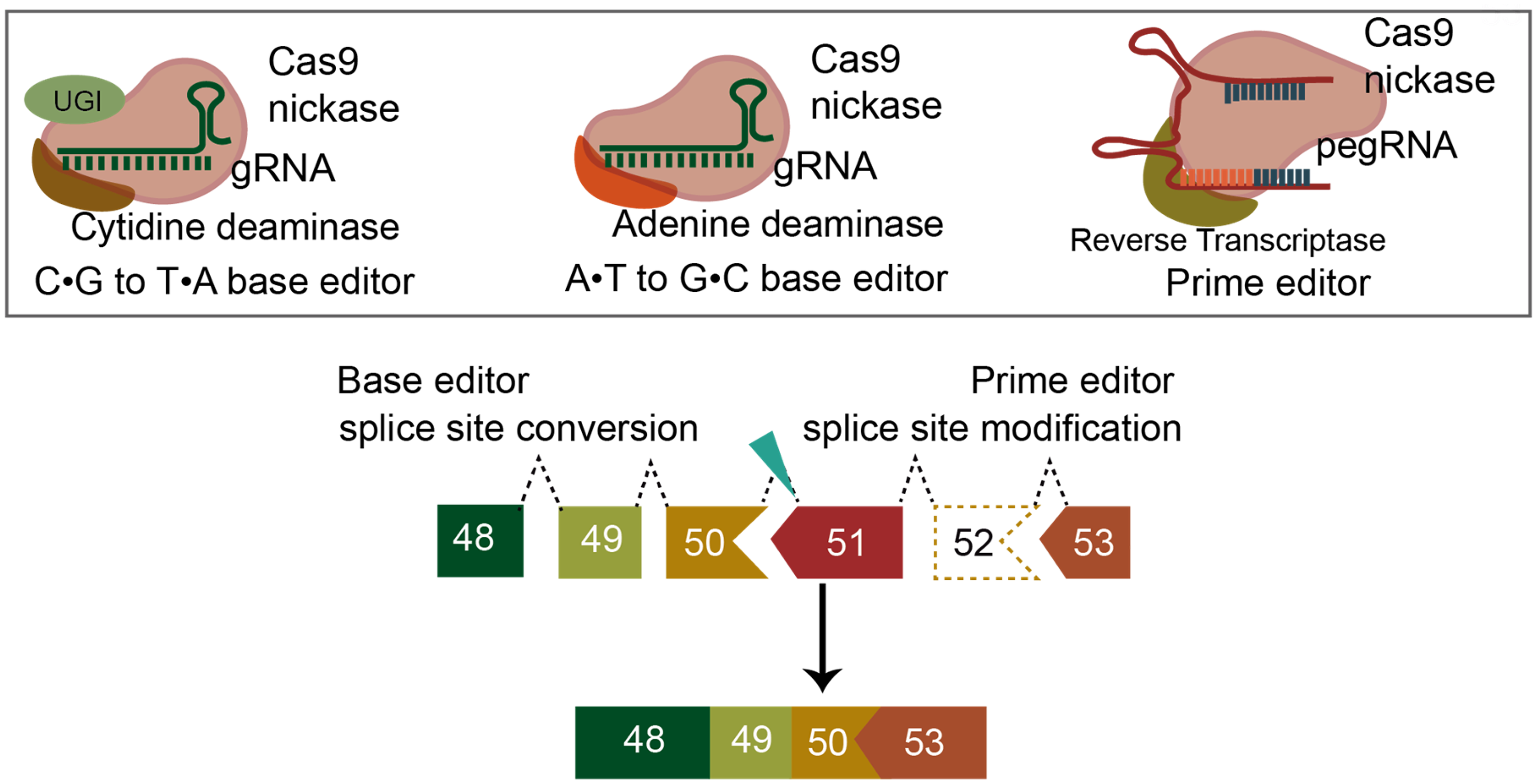
In vivo application of BE technology posed challenges due to its size exceeding the AAV packaging capacity. However, several groups have devised solutions, including delivering BE components via dual trans-splicing AAV vectors and split-intein-mediated splicing AAV vectors.63,64 These delivery strategies have been rapidly adopted by other research teams. Chemello et al. employed split-intein ABE AAV delivery to skip exon 50, resulting in 35% genome editing locally, with 96% of myofibers showing restored dystrophin. 60 Gapinske et al. also used the split-intein strategy for ABE delivery. Using two different capsids, AAV9 and myoAAV, they achieved successful ABE administration both intramuscularly (AAV9) and systemically (myoAAV) in wild-type mice. Intramuscular injection with AAV9 yielded a 20% editing rate, while systemic injection with myoAAV resulted in generally low editing rates in muscle tissue, with a median of 7.6% observed in cardiomyocytes. 65 Another strategy to overcome the limited AAV capacity is using the small orthologs of Cas9 to develop new BEs, such as Campylobacter jejuni (2.95 kb), Neisseria meningitidis (3.24 kb), and Staphylococcus auricularis (3.18 kb).66–68 A recent evolution of BE, ABE8e has further minimized the size of the editor and has been tested with those three orthologs. 69
PE is the latest advancement of the non-DSB approach in CRISPR technology. It has greater versatility compared to base editors, allowing for various DNA alterations, including substitutions, insertions, and deletions of nucleotides or sequences. PE has demonstrated efficacy in vitro for inducing exon skipping by modifying splice donor sites for exon 51 or exon 53 to skip single or multiple exons60,70 (Figure 4). Similar to BE, its large size presents a challenge for in vivo delivery using AAV vectors. While Davis et al. have developed a method to split PE into two AAV vectors and fuse them using fast-splicing intein, PE in vivo has not been applied to induce exon skipping in DMD to date. 71 The development of PE has enabled RNA-guided DNA integration with genome editing tools including PASTE, that, in theory, could enable exon replacement. The next generation of precise editors including PE, PASTE, type I CRISPR-associated transposons and DNA-dependent DNA polymerases may enable precise correction of the DMD gene.72–74
The specific protospacer adjacent motif (PAM) requirements (NGG for SpCas9 or NNGRRT for SaCas9) limit the genomic targets accessible by these enzymes. Researchers have identified several Cas9 alternatives and are exploring their use in genome editing approaches. 75 Cas12a is the second most commonly used Cas9 alternative. The PAM sequence of Cas12a (TTTN or TTN) differs from that of SpCas9 and SaCas9 and has greater flexibility in recognition sequence in AT-rich regions, allowing it to target new genomic locations. Moreover, Cas12a size is generally smaller than Cas9, and its guide length is shorter than that of guide sequence of Cas9. 76 Multiple research groups have demonstrated Cas12a's effectiveness in skipping single or multiple exons to restore DMD gene expression in cells derived from individuals with DMD.77,78 Further, they showed that the editing efficiency of Cas12a is comparable to that of Cas9.
Another promising class of Cas enzymes for editing technology is Cas3, known for its efficient capability to generate large genomic deletions. 79 Morisaka et al. pioneered the application of the Cas3 enzyme in mammalian genome editing and DMD gene correction, successfully restoring dystrophin expression in DMD-iPSC cells through exon 45 skipping. 80 Additionally, Kita et al. were the first to employ the dual CRISPR-Cas3 system to induce a large deletion (∼340 kb) in the DMD gene exon 45–55 region. 81
Delivery challenges and consideration
Adeno-associated virus (AAV) is widely used in gene therapy for DMD due to its innate ability to target muscle tissue effectively.82,83 Current AAV serotypes used for systemic gene therapies targeting muscles in the clinic have an intrinsic tropism for muscles are AAV8, AAV9, and AAVrh74.84–86 Although these serotypes aim to target skeletal and cardiac muscles, they also remain heavily targeting the liver due to their lack of selectivity. Further, due to its distribution and mass in the body, effectively editing all target muscles requires administering very high doses of these serotypes systemically. This approach has not only resulted in increased costs but has also led to the emergence of severe toxicities as observed in a patient with DMD treated with high-dose rAAV gene therapy. 87
An optimal AAV vector should express the therapeutic transgene product at high levels specifically and efficiently in the desired target tissue. Two muscle-targeting vectors, known as AAVMYO or MyoAAV reported by Weinmann et al. (2020) and Tabebordar et al. (2021) respectively, have demonstrated remarkable success in achieving better muscle tropism with lower doses.88–90 These vectors share the characteristic display of the amino acid motif RGD (arginine–glycine–aspartate) on their capsid surface and demonstrate the most effective transduction rates in skeletal muscles of mice or nonhuman primates documented thus far. Reaching muscle satellite cells is paramount to long-term mainanence of gene therapy. While AAV transduces skeletal muscle efficiently, satellite cell transduction has been limited and difficult to measure. 91 Previously, Kwon et al. demonstrated that AAV8 and AAV9 show the highest tropism for satellite cells (defined by Pax7-nGFP expression) and sustained dystrophin expression in AAV9-CRISPR treated mice after serial muscle injuries. 92 Tabebordar et al. showed, using MyoAAV 1A, transduction efficiency in satellite cells (defined by CD29 and CD184) was 2.6 and 3.2 times higher compared to AAV9 and AAV8, respectively. 88 More recently, Zengel et al. showed an increase in satellite cell tropism (defined by a7+/CD34+/CD45-/CD11b-/CD31-) by overexpression of AAVR showing between 4-fold and 36-fold improvements in delivery. 93
While promising, the evaluation of AAVMYO, MyoAAV, and future capsids remains paramount. It is unknown whether the production and packaging of these vectors will be efficient at the required clinical scale while maintaining stability and potency. Furthermore, it is uncertain whether natural AAV seropositivity will impede dosing with AAVMYO or MyoAAV in humans. Additionally, there is a need to assess the influence of the AAVMYO or MyoAAV capsid on the epigenetic status of the vector genome, as reports suggest that the AAV capsid protein impacts the epigenetic status of the vector genome, leading to transgene silencing.94,95
AAV delivery poses a challenge in terms of repeated administration of the same serotype due to AAV neutralizing antibody. An alternative approach involves using non-viral delivery methods, such as lipid nanoparticles (LNP). However, studies on CRISPR delivery in skeletal muscle using LNPs are limited, primarily due to their limited systemic efficiency compared to AAV. Nevertheless, Kenjo, et al. developed a new formulation of LNP delivery system designed to target skeletal muscle. 96 This LNP system has shown results in inducing exon skipping to restore dystrophin levels in DMD mice. Importantly, the LNP exhibited sustained luciferase expression following multiple intramuscular injections when compared to AAV.
Porous silicon nanoparticles (PSiNPs) are another attractive non-viral drug carrier due to their loading capacity, which is >10 fold higher than other systems, making them a potential solution for limited AAV capacity. 97 It has also been reported that PSiNPs are non-toxic and biodegradable. 98 Fletcher et al. reported the application of PSiNPs for Cas9 mRNA/gRNA complex delivery. Using a BaCl2 muscle injury model, they achieved 2% editing in the DMD gene after intravenous administration. 99 This level of editing is comparable to the early AAV-CRISPR mediated exon skipping studies in DMD. Despite efforts in improving editing efficiency with novel muscle-tropic capsids or LNPs, only a fraction of myonuclei per myofiber is edited. Successful CRISPR-Cas9-based genome editing in skeletal muscle relies on efficient Cas9 delivery to all myonuclei. Factors such as Cas9 protein size and nuclear localization sequence length limit propagation efficiency, leading to poor editing efficacy. Poukalov et al. identified Myospreader, a short peptide sequence that enhances myonuclear propagation, stabilizes Cas9 protein, and increases muscle gene editing efficiency in vivo. 100
Another challenge in delivery is the immune-related challenges to the Cas9 protein administration. Prior exposure to Cas proteins from specific bacterial species may trigger an immune response, potentially resulting in the clearance of Cas endonucleases and compromising treatment efficacy. 101 Studies have revealed that a substantial percentage of individuals harbor anti-Cas9 antibodies, highlighting a significant risk of immune response following Cas9 treatment.102–104 The first demonstration of AAV-delivered CRISPR in a canine model of DMD by Amoasii et al. showed no evidence of mononuclear cell infiltration after intramuscular injections with transient immunosuppression. In contrast, Hakim et al. found that the Cas9-immune responses present a major challenge for AAV CRISPR therapy in canines. 105 Their research showed restoration of dystrophin in canine DMD models using AAV-CRISPR through local and systemic injections. However, they also noted that the expression of Cas9 triggers a humoral response and cytotoxic T lymphocytes in wild-type and DMD dogs leading to the loss of CRISPR-rescued dystrophin in treated dogs. It is also crucial to highlight that conventional methods like prednisolone or tissue specific promoters were insufficient in managing Cas9-immune response in this study. More research is needed to understand the long-term consequence with emerging vectors, Cas9 orthologs, and new muscle-specific promoters.
Various strategies have emerged to mitigate the immune response to Cas9 protein. These include adoptively transferring Cas-reactive Treg cells, immunosilencing proteins by concealing immunogenic epitopes, and using immune orthogonal CRISPR effector orthologs for repeated dosing.104,106,107 Furthermore, the timing of treatment proves critical, as neonatal mouse models exhibit the capacity to reduce both humoral and cellular immune responses after CRISPR-DMD treatment. 108
Future perspectives and conclusions
As a disease characterized by a spectrum of mutations, targeted gene integration methods have emerged as treatments for DMD, aiming for mutation-independent solutions. This approach enables the restoration of full-length dystrophin instead of producing a partially functional protein, as seen in BMD. While delivering a healthy DMD transcript remains a challenge with current delivery methods, recent studies by Pickar-Oliver et al. (2021) and Stephenson et al. (2023) have proposed strategies involving the delivery of superexon transcripts (exon 51–79) or mega-exon transcripts (exon 1–19) to achieve full-length DMD restoration.109,110 By employing homology-independent targeted integration (HITI), simultaneous DSBs in the genome and a donor vector containing the superexon guide insertion of the vector. In each case, they successfully demonstrated the expression of full-length human DMD 91 or mouse Dmd 90 in mouse models.
Although promising, similar to DSB mediated exon skipping, concerns arise with DSB mediated integration due to the levels of AAV vector integrations observed in both studies on DMD exon integration, involving vector fragments such as the ITR, portions of the Cas9 reading frame, or gRNAs. A new approach involves exploring DSB mediated integration through the combination of CRISPR associated transposons or a fusion of prime editor with integrases offering a scarless method for DNA integration.72,74,111,112 However there have been no studies testing these methods on the DMD gene yet.
Until now, exon skipping remains the most viable and widely used method for genome editing, primarily due to the current delivery options, especially the AAV capacity. Various CRISPR-mediated exon skipping methods have effectively been used in both DMD cells and animal models, showing potential for treating DMD. While challenges exist in delivering treatments and managing immune responses, there are global efforts to tackle these challenges. In addition to the exon skipping strategy, progress in gene integration approaches is also promising. Together, these advancements are paving the way for the next generation of genome editing therapies, bringing hope for the development of long-lasting treatments for DMD.
Footnotes
Acknowledgments
This work was supported by the NIH/NIBIB under Grant No. R00EB023979, The American Society for Gene and Cell Therapy Career Development Award, The University of Arkansas Chancellors Innovation Fellowship, and the Arkansas Bioscience Institute. CEN was supported by the twenty-first Century Chair in Biomedical Engineering. MHP was supported by the Fulbright Indonesia Research Science and Technology (First) Master's Degree Program and PhRMA Foundation 2023 Predoctoral Fellowship in Drug Discovery. SA was supported by a University of Arkansas Women’s Giving Circle Grant.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
MHP and CEN are named inventors on patents and patent applications related to genome editing.
Correction (June 2025):
The article has been updated to correct the article type from “case report” to “invited review”.