
Abstract
Background:
Paramyotonia congenita (PC; OMIM 168300) is a non-dystrophic myotonia caused by mutations in the SCN4A gene. Transient muscle stiffness, usually induced by exposure to cold and aggravated by exercise, is the predominant clinical symptom, and interictal persistent weakness is uncommon.
Case report:
We report a family with a history of PC accompanied by persistent hand muscle weakness with masticatory muscle involvement. Persistent weakness was exacerbated with age, and MR analysis showed marked atrophy of temporal, masseter, and finger flexor muscles with fatty replacement. The PC causative mutation T1313M in the SCN4A gene was prevalent in the family. Administration of acetazolamide chloride improved clinical symptoms and the results of cold and short exercise tests. Phenotypic variation within the family was remarkable, as the two younger affected patients did not present with persistent weakness or muscle atrophy.
Conclusions:
PC associated with the T1313M mutation is a possible cause of persistent distal hand weakness.
Keywords
LIST OF ABBREVIATIONS
Paramyotonia congenita sodium channel protein type 4 subunit alpha compound motor action potential electrocardiogram echocardiogram
BACKGROUND
Paramyotonia congenita (PC, OMIM 168300) is a non-dystrophic myotonia caused by mutations in the sodium channel protein type 4 alpha sub-unit (hNaV1.4), which is encoded by the SCN4A gene [1]. A hallmark of the PC phenotype is the marked exacerbation of muscle rigidity upon exposure to cold (paramyotonia). Clinical symptoms of PC are usually present during early childhood and often remain unchanged for life, and muscle atrophy is not typical for the disease [2].
Although several case reports have reported patients who presented with mild weakness [3–15], these weaknesses are mild and proximal dominant. Here, we present the detailed phenotype of a Japanese family with a history of PC accompanied by severe distal weakness together with masticatory muscle involvement. This is the first report, to our knowledge, that provides detailed muscle images revealing the myopathic nature of the weakness in PC patients. Furthermore, we performed a literature review on previously reported cases of PC, including only those in which genetic analysis was performed and clinical information was provided. Written consent for publication was obtained from all patients.
CASE PRESENTATION
Case 1 (III-2)
The proband had no developmental abnormalities. Since childhood, she had difficulty gripping horizontal bars, and had trouble writing during the winter and on days with cold weather (or with a sudden drop of temperature), which she had no problems with during the summer and on days with warm weather. During elementary school, she noticed a feeling of weakness whenever she suddenly used her muscles, felt stiffness of muscles after running 30 m full power in a 50 m short track sprint, and was unable to keep running after a 50 m sprint. She also had difficulty chewing and became aware of fatigue upon chewing firm food. In her 40s, she had difficulty opening pull-tabs, even on days with hot weather. She had noticed that her grip power was weaker than others. When finger weakness worsened, she visited our hospital at age 61. She recalled that her mother (case II-2) experienced similar finger weakness and muscle stiffness. She did not experience episodic paralysis.
On examination, she presented with right strabismus, and her extraocular movements were limited except for left extension. She had paramyotonia of the eyelids and masseter muscle. Her temporal and masseter muscles were atrophic and a narrow high-arched palate was observed. No tongue atrophy was noted. The manual muscle test revealed persistent distal dominant weakness (Table 1). Grip power was 1/1 kg, forearm and intrinsic muscles were atrophic, and the proximal interphalangeal joint was hyperextended. Although she had mild weakness of proximal muscles, her shoulder muscles or lower limb muscles were mildly hypertrophic. Deep tendon reflexes were normal, and there were no other abnormalities. CK was 127 IU/L (within normal range). Needle electromyogram revealed myopathic changes together with abundant myotonic discharges in all muscles examined. We performed repeated short exercise and cooling tests to stimulate the ulnar nerve [16–18], as well as additional longer stimulation to observe prolonged recovery. These tests revealed marked decrement of compound muscle action potential (CMAP) after exercise to 20% (1.10 mV) of the baseline value (5.59 mV), with subsequent partial recovery (Fig. 2A). The cooling test also revealed marked decrement of CMAP to 4% (0.36 mV) of the baseline value (8.96 mV) (Fig. 2B). Muscle CT revealed mild hypertrophy of proximal muscles, and low density and atrophy of the forearms (Fig. 3). Forearm MRI revealed fatty changes and atrophy of forearm muscles, especially the flexor digitorum profundus, flexor digitorum superficialis, extensor pollicis index and longus, supinator, and adductor pollicis longus muscles, whereas flexor carpi radialis and flexor carpi ulnaris muscles were relatively spared (Fig. 4A–E). Cranial MRI revealed temporal and masseter muscle atrophy with fatty replacement (Fig. 4F–H). There were no abnormal findings on ECG, UCG, holter ECG, pulmonary function tests, nocturnal respiratory monitoring, and videofluorography. No abnormalities were found in the DMPK and CNBP genes. However, analysis of the SCN4A gene revealed a C to T mutation at c. 3938 in exon 22 (p.T1313M). No additional abnormalities were found in genomic DNA isolated from peripheral blood, examined by whole-exome sequencing using Hiseq1000 (Illumina) and sequence data analyses as described previously [19].
Summary and characteristics of presented cases (Patients 1 (III-2), 2 (III-1), and 3 (IV-5))
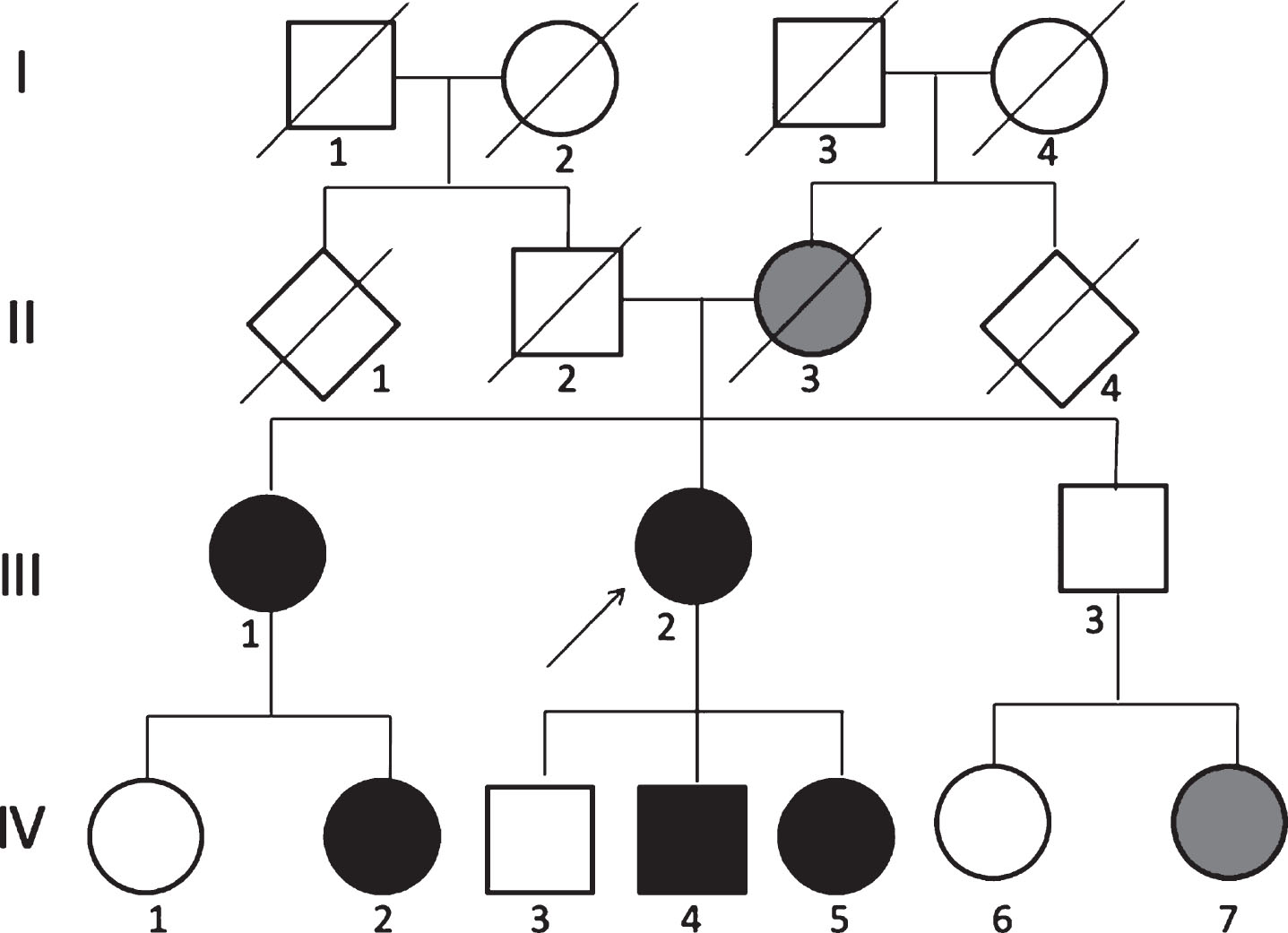
Family tree. Cases represented by black circles were examined by neurologists and diagnosed with paramyotonia. Cases II-2 and IV-7 (gray) were not diagnosed but exhibited similar symptoms as Cases III-1 and III-2, and thus were suspected of having paramyotonia.
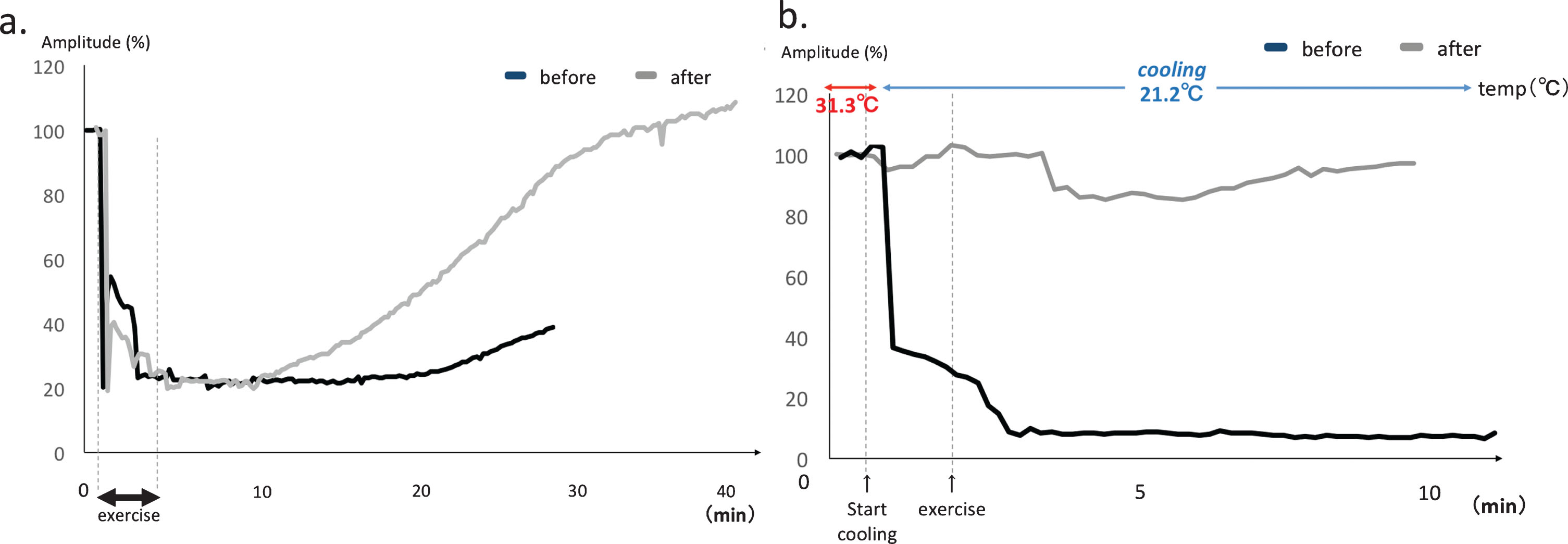
Electrophysiological tests on Case III-2. Black line = baseline (before treatment), gray line: after acetazolamide treatment. (A) Short exercise test of right ADM. Marked decrement after exercise to 20% (1.10 mV) of baseline CMAP (5.59 mV, black line), and partial recovery was observed thereafter (gray line). (B) The cooling test also showed marked decrement to 4% (0.36 mV) of baseline CMAP (8.96 mV, black line), whereas no significant decrement was observed after treatment (gray line).
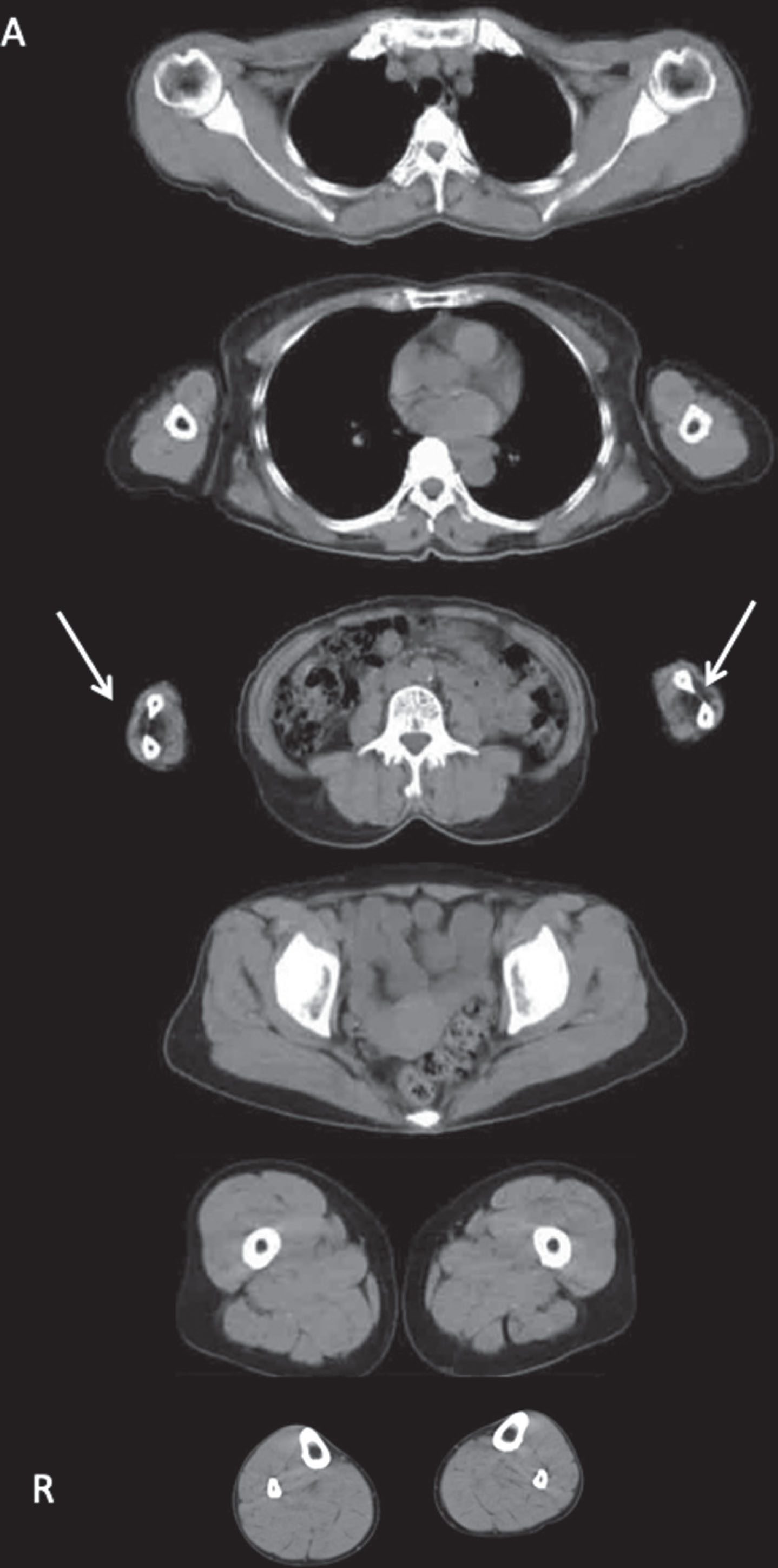
Muscle CT of Case 2 (III-2). CT showed mild hypertrophy of proximal muscles, with the exception of atrophy and low density of forearm muscles (arrow).
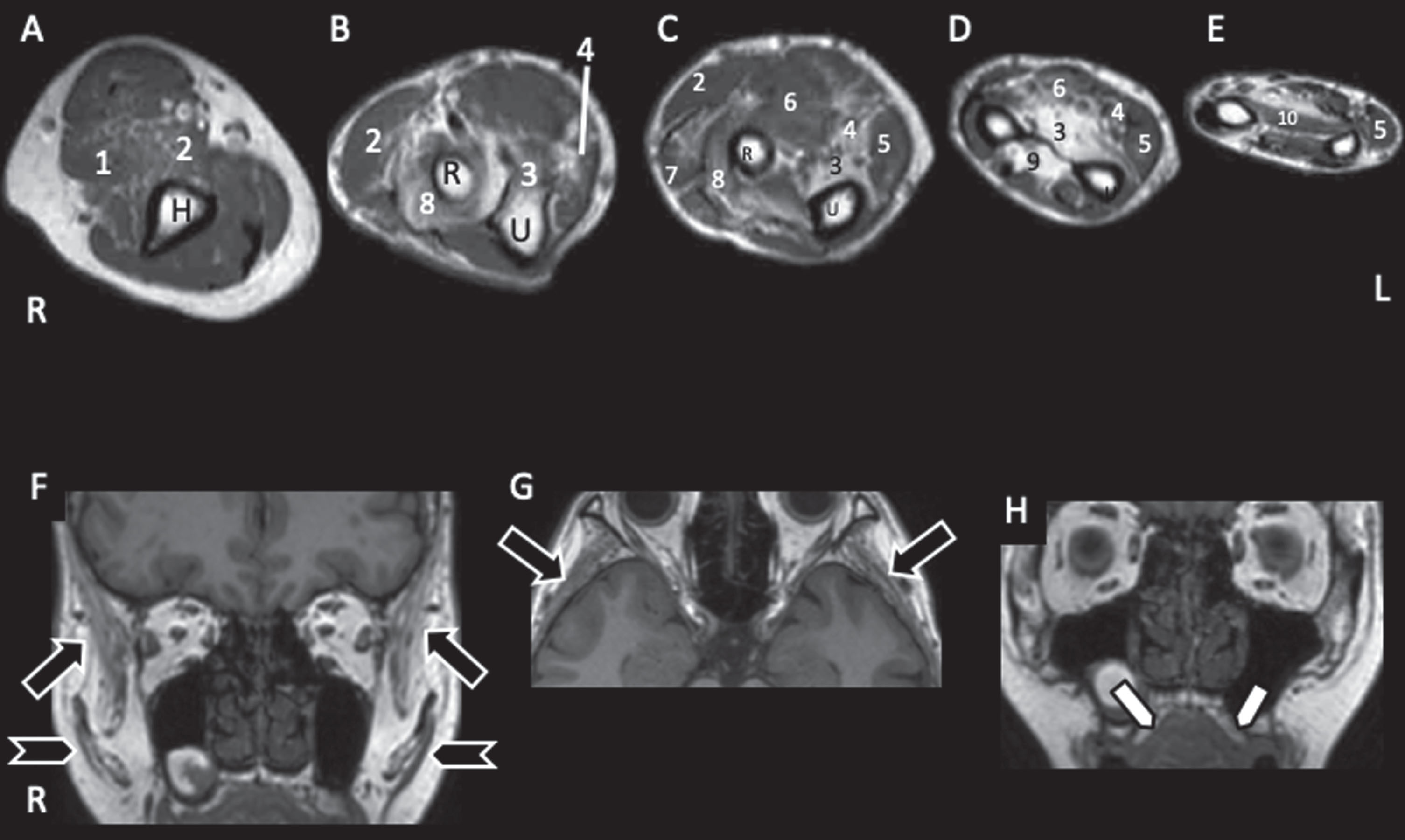
Muscle and Cranial T1WI MRI of Case 2 (III-2). A; right upper arm, B–E; forearm, F–H; cranial images. U: ulna, R: radius, 1; brachialis, 2; brachioradialis, 3: flexor digitorum profundus (FDP), 4: flexor digitorum superficialis (FDS), 5: flexor carpi ulnaris (FCU), 6: flexor carpi radialis (FCR), 7: extensor carpi radialis (ECR), 8: supinatus, 9: abductor pollicis longus (APL), 10: pronator quadratus (PQ). A: Upper arm muscles were well-preserved, although mild fatty infiltration was observed in brachialis (3) and brachioradialis (2) muscles. B–D: FDP (3), FDS (4), supinatus (8), and ALP (9) showed marked fatty changes and atrophy, whereas FCU (5), FCR (6), and ECR (7) were relatively spared. These findings are compatible with marked finger weakness and loss of grip power. F, G: Temporal muscles (arrows) and masseter muscles (arrow heads) showed marked atrophy and fatty infiltration. H: Arched palate (arrow) was observed.
Administration of 500 mg acetazolamide improved her paramyotonia except for the persistent weakness. The time needed for 10 serial handgrips and 10 serial squats improved from 29.1 seconds to 13.9 seconds and 29.3 seconds to 17.2 seconds, respectively. Serial grip power measurements also improved from 15% (2 kg to 0.3 kg) to 88% (2.5 kg to 2.2 kg). In short exercise and cooling tests, CMAP increased relative to before drug administration, and CMAP decrement after exercise also improved with drug administration (Figures 2A and 2B). Her younger son (Case IV-4) also tried acetazolamide and felt that it was effective.
Case 2 (III-1)
The patient had poor physical performance in her school days, and she was aware of hand weakness. She could not grip horizontal bars or a tennis racket to the end of a game. She felt tired when chewing tough meat. These symptoms worsened in the winter. She did not experience episodic paralysis. She visited a neurologist in her late 40 s and was diagnosed with myopathy following nEGM examination. The administration of 150 mg mexiletine hydrochloride was started but did not improve her symptoms. Acetazolamide was never tried. When she visited the neurologist again at age 63, she presented with facial muscle involvement and temporal muscle atrophy, as well as distal arm muscle atrophy, persistent weakness, and grip myotonia that worsened with recurrent grips. She had no difficulty walking. Deep tendon reflexes were reduced in the arms, but no other abnormalities were observed. EMG revealed abundant myotonic discharges. Her daughter (IV-2) had similar persistent weakness and paramyotonia.
CASE 3 (IV-5)
The patient had difficulty fastening buttons and writing during the winter since childhood. She also noticed that repeated handgrips resulted in weakness. She was a slow runner and had poor physical performance in her school days. She felt her legs became very “stiff,” like clubs, and could not run in cold temperature conditions. She did not experience episodic paralysis. She visited a neurologist at age 24. On examination, she presented with grip and eyelid paramyotonia. No weakness or muscle atrophy was noted. Deep tendon reflexes were normal. Administration of 250 mg acetazolamide chloride did not improve her quality of life. Her brother (IV-4) had similar symptoms.
DISCUSSION AND CONCLUSION
This is the first report of significant persistent distal and cranial muscle weakness and atrophy identified by muscle imaging in patients with PC. Most reports on persistent weakness focused on proximal weakness, whereas our cases and another Japanese study [6] reported distal weakness among patients harboring a T1313M mutation in the SCN4A gene. Among the three affected members in the Japanese study, two had mild grip weakness [6].
We reviewed case reports that mentioned neurological examinations of clinically diagnosed PC patients (i.e., did not contain borderline symptoms of other phenotypes) with mutations in the SCN4A gene, and identified reports describing those positive for persistent or fixed weakness in the interictal phase. Patients described as having “no persistent/fixed weakness” or “no weakness,” or “no abnormal findings in neurological examination” were considered “no persistent weakness” cases. In the 49 papers analyzed (more than 89 pedigrees, 256 cases; Supplementary Table 1), 46 patients (18%) had persistent weakness [3–15] (Table 2), 126 (49%) had no persistent weakness, and 84 (32%) had no description about weakness. Among those with persistent weakness, 8/46 (17%) were aged under 30 years, 17/46 (37%) were aged 30 years and above, and age was unknown in 21/46 (46%). Of the 42 patients with persistent weakness, 16 had proximal weakness [3–5,8, 13], five had mild distal hand weakness [6, 10], and 26 had no description regarding weakness distribution [7, 9]. Among 29 patients carrying the SCN4A T1313M mutation described in the 12 studies (more than 22 pedigrees; Table 3), 4/50 had persistent weakness [6, 14], 16/50 had no weakness, and 30/50 had no description about weakenss. One unique feature of our cases was the presence of both proximal and marked distal weakness.
Summary of permanent weakness in genetically confirmed PC cases
The marked persistent weakness, muscle atrophy, and fatty changes observed in our cases reveal the myopathic nature of the disease. As Bednardz suggested [13], the pathogenesis of sodium-channel myopathies may involve increased sodium influx and osmotic edema that strangulate T-tubules, leading to fiber necrosis and stimulation of new T-tubule proliferation, vacuolar formation [21, 22], and hyperosmotic stress with mitochondrial degeneration [23]. Persistent weakness was more prevalent among patients aged over 30 years than those under 30 years (Tables 2 and 3). Recurrent myofiber stress may be responsible for fiber damage due to necrosis. It remains unclear whether recurrent paramyotonia increases the risk of developing weakness, although it is noteworthy that permanent weakness was evident in muscles which experienced recurrent paramyotonia, and that the onset of weakness was evident after paramyotonia appeared. We found 3 reports comprising 8 cases that mentioned muscle biopsy, including a T1313M mutation case series. Of the eight cases biopsied, normal pathological results were observed in 4 cases, while vacuolar changes, tubular aggregates, and scattered regenerating fibers were observed in 1 case each [2, 64]. One biopsy ([2]) showed scattered regenerating changes, indicating a necrotic-regenerating process that can cause permanent myopathic changes.
In this study, we were able to detect both muscle atrophy and fatty replacement by MRI and CT imaging. An ultrasound study of 19 sodium channelopathy cases, including 10 PC cases, revealed increased echointensity in the biceps brachii and forearm flexors [24]. Our study provides further detail by showing that flexor digitorum superficialis and flexor digitorum profundus muscles were strongly involved among forearm flexors. These findings are compatible with the observations of marked finger weakness, facial muscle atrophy, and fatty changes. Moreover, muscles with severe transient weakness, such as finger, facial, and masseter muscles, were markedly myopathic among muscles.
Case III-2 is remarkable in that it is the first case of PC in which the patient presented with strabismus and an arched palate. A few reports have described strabismus among SCN4A-related myotonia cases [9, 25]. Strabismus and/or diplopia was reported in 47% of children with SCN4A-related myotonia among those with sodium channelopathies [9], indicating an overlap in phenotype. Previous reports on facial involvement and arched palate included a myopathy patient with a recessive loss-of-function mutation who did not present with myotonic features of those carrying SCN4A mutations [26, 27]. Severe facial muscle involvement may potentially explain the arched palate.
The paramyotonia observed in one of our patients improved with acetazolamide chloride and, as with previous reports [28–30], we described the electrophysiological interval changes in this patient. However, mexiletinene, which has been suggested to reduce the sensitivity of the mutated sodium channel to mexiletine [31] and reportedly has beneficial effects in T1313M mutation carriers [28, 32], had no effect in the one affected patient who was treated with the drug.
In conclusion, PC patients harboring SCN4A mutations can present with persistent weakness of distal hand and cranial muscles. Phenotype varies even within a family and with age. Therapeutic approaches may counter the progression of persistent weakness.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
Footnotes
ACKNOWLEDGMENTS
This study was supported partly by an Intramural Research Grant (23-4, 26-7, 28-6, and 29-4) for Neurological and Psychiatric Disorders of NCNP, Research and Development Grants for Practical Research Project for Rare/Intractable Diseases (16ek0109066h0003), and Research Grants for Nervous and Mental Disorders (20-11) from the Ministry of Health, Labour and Welfare of Japan. The role of the funding bodies was restricted to providing funding support to the first author for data collection and drafting the manuscript.