
Abstract
Spinal Muscular Atrophy (SMA) is caused by autosomal recessive mutations in SMN1 and results in the loss of motor neurons and progressive muscle weakness. The spectrum of disease severity ranges from early onset with respiratory failure during the first months of life to a mild, adult-onset type with slow rate of progression. Over the past decade, new treatment options such as splicing modulation of SMN2 and SMN1 gene replacement by gene therapy have been developed. First drugs have been approved for treatment of patients with SMA and if initiated early they can significantly modify the natural course of the disease. As a consequence, newborn screening for SMA is explored and implemented in an increasing number of countries. However, available evidence for these new treatments is often limited to a small spectrum of patients concerning age and disease stage. In this review we provide an overview of available and emerging therapies for spinal muscular atrophy and we discuss new phenotypes and associated challenges in clinical care. Collection of real-world data with standardized outcome measures will be essential to improve both the understanding of treatment effects in patients of all SMA subtypes and the basis for clinical decision-making in SMA.
Keywords
INTRODUCTION
The SMA landscape has changed considerably since the first reports more than a century ago of patients with spinal muscular atrophy (SMA) by Werdnig and Hofmann in 1891 [1] and 1893 [2]. Decoding the disease’s genetic background, first in linkage analyses [3, 4] and later by identifying mutations in SMN1 as disease-causing [5], paved the way for targeted medical approaches. In this review we provide an overview of both the latest therapeutic options and emerging therapies for SMA. We also discuss new topics and challenges arising with the availability of drug treatments that alter the known trajectories of disease. These include changing phenotypes, new medical decisions, and newborn screening for SMA.
BACKGROUND
SMA is one of the most frequent monogenic neurodegenerative diseases with an incidence estimated to be around 1 : 6,000 to 1 : 10,000 in newborns [6–9]. SMA encompasses a wide clinical continuum of disease severity and has been classified into subtypes according to age at onset and the motor milestones achieved [10]. More than half of patients have the severe phenotype of SMA type 1 with onset of symptoms within the first 6 months of age. A ‘floppy infant’ presentation, reduced spontaneous movements and a paradoxical breathing pattern are characteristic; these infants fail to achieve the free-sitting milestone. Without drug treatment and ventilator support, SMA type 1 is the leading genetic cause of death in early infancy with a life expectancy of under 2 years [11, 12]. SMA type 2 is characterized by a milder course with onset of symptoms between the ages of 6 and 18 months. Per definition, these patients do manage free sitting, but not independent walking. The latter is achieved (at least temporarily) in patients with SMA type 3, whose symptoms’ onset is during infancy or adolescence. In addition, some classifications define SMA type 0 and SMA type 4 with prenatal onset or a very mild phenotype entailing an adult onset of symptoms, respectively. The disease’s hallmark is the degeneration of anterior horn cells in the spinal cord, leading to the characteristic symptom of progressive, proximal weakness involving varying degrees of muscle atrophy. Whereas all types of SMA are progressive, the rate of progression differs: SMA type 1 typically follows a rapidly progressing course, while type 3 progresses slowly [13].
Molecular genetics: About 95% of SMA cases are caused by homozygous deletions and less frequently point mutations in the SMN1 gene (
In patients with SMA on the other side, small amounts of full-length and fully functional SMN-protein can be produced by SMN2 [20], thus higher numbers of SMN2-copies are associated with milder phenotypes [21, 22]. Figure 1 summarizes SMA subtypes and displays typically associated SMN2 copy numbers.
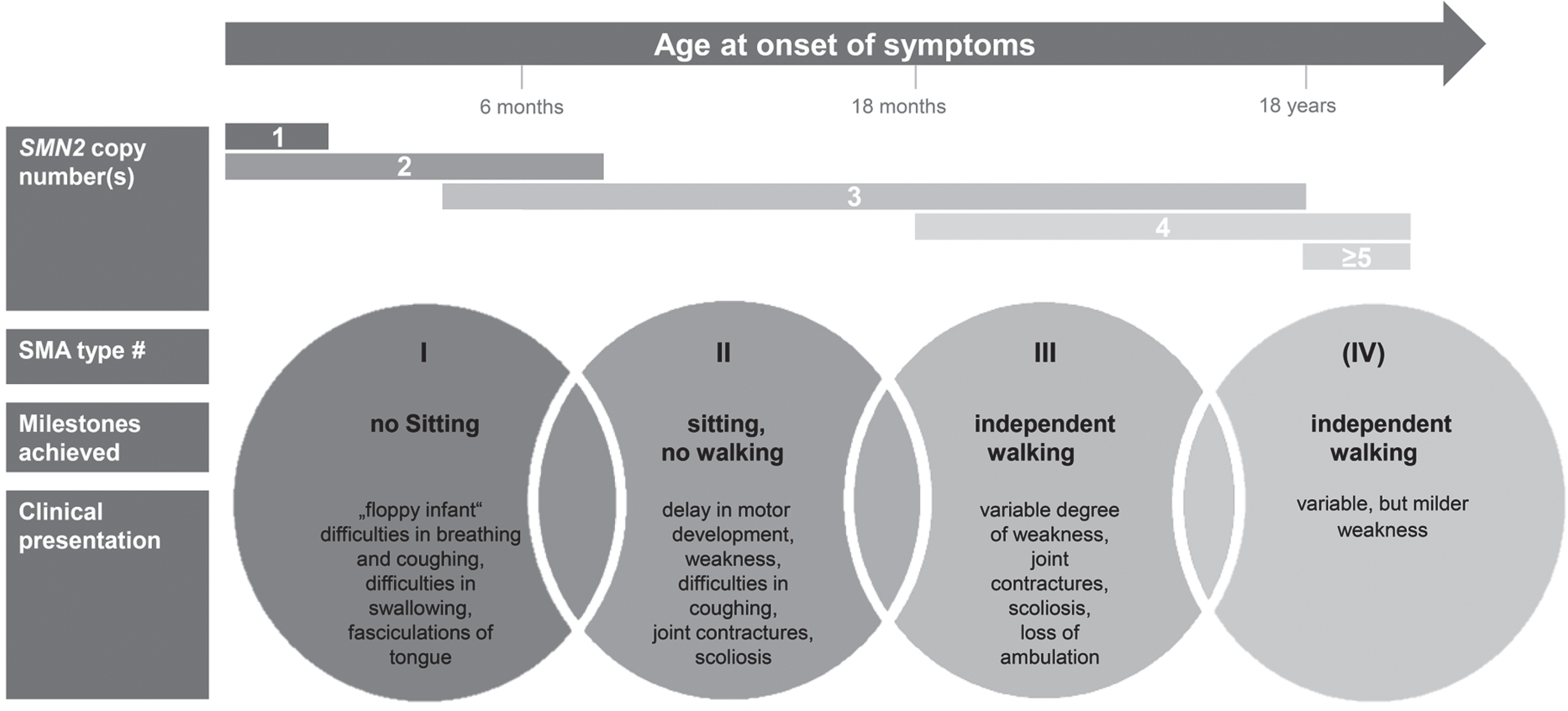
Clinical classification of SMA subtypes according to onset, milestones achieved, and clinical presentation. Typically associated SMN2 copy numbers are displayed.
THERAPEUTIC APPROACHES – SYMPTOMATIC TREATMENT
While being a monogenetic neuromuscular disease, the resulting phenotypic spectrum is complex and SMA is generally perceived as a systemic disease [23]. Accordingly, caring for patients with SMA requires the interdisciplinary management of respiratory, nutritional and gastroenterological, orthopedic, and psychosocial issues. General treatment recommendations were published in 2007 in the first consensus statement on standards of care in SMA [24]. Nevertheless, the implementation of standards of care is highly variable and is influenced by cultural perspectives, socioeconomic factors, and the availability of regional resources [25]. Due to advances and improvements in care over the last decade, an updated version of recommendations on diagnosing SMA and patient care was published only recently [26, 27].
THERAPEUTIC APPROACHES – DRUG TREATMENT
Several different compounds have been investigated in randomized controlled trials in the last few decades, including approaches to increase muscle strength and function by (1) hyperacetylating agents such as valproic acid [28–30] or phenylbutyrate [31], (2) anabolic agents such as albuterol [32], thyreotropin-releasing hormone [33] or growth-hormone [34] and (3) neuroprotective agents such as gabapentin [35, 36], riluzol [37] and olesoxime [38]. Despite negative results regarding primary endpoints, those investigations validated outcome measures and yielded key information about trial designs and the feasibility of patient recruitment.
Actual therapeutic developments can be subdivided into therapies aiming to modify the splicing of SMN2, replacing the SMN1 gene, or upregulating muscle growth. Figure 2 summarizes the therapeutic approaches discussed in the following sections and illustrates the respective molecular mechanisms of action; Table 1 illustrates the current status of development of specific drugs.
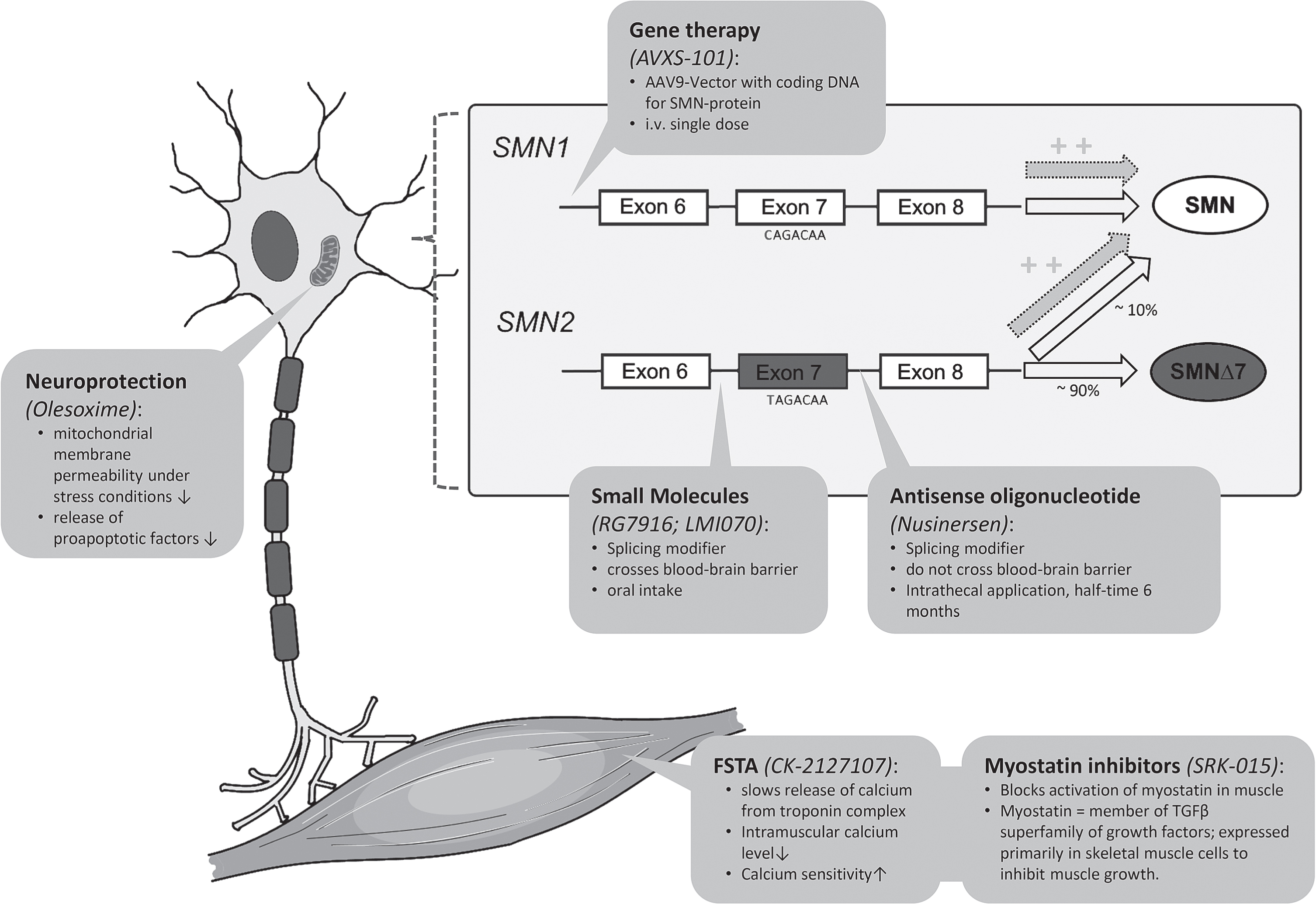
Synopsis of selected ongoing and recently finished clinical trials of medical treatments in spinal muscular atrophy (SMA). AAV-9 = Associated Adenovirus 9; 6MWT = six minute walking test; FSTA = Fast Skeletal Muscle Troponin Activator; IT = intrathecal; PO = oral intake; IV = intravenous application
Splicing modification of SMN2
The first drug approved for SMA treatment was nusinersen (former IONIS-SMNRX), an
An approach to altering the splicing of SMN2 and thus increasing the amount of functional SMN-protein is also taken by
Replacement of SMN1-gene
Upregulation of muscle growth
Therapeutic approaches that do not directly target the genetic cause of SMA include the improvement of muscle mass and function. Two compounds are the most advanced:
Therapeutic approaches in preclinical development
A growing number of compounds are in preclinical or early clinical development. Those comprise small molecules aiming to stabilize the SMN-protein and other types of ASOs targeting SMN2, but also SMN-independent approaches. The latter include myostatin-inhibition via Activin Receptor Type IIB-antagonists [60] and stand-alone approaches like inhibition of the p38MAPK pathway [61].
BIOMARKERS IN SMA
Apart from SMN2 copy numbers, a variety of other possible biomarkers are currently discussed and investigated [62]. Within the ENDEAR study population, symptomatic SMA type 1 patients showed higher levels of plasma phosphorylated neurofilament heavy chain (pNF-H) than healthy controls. Furthermore, higher pNF-H levels correlated positively with earlier onset of symptoms and inversely with motor function at start of nusinersen treatment. Under treatment with nusinersen, these levels decreased faster in the verum group than in sham control group. This decrease was more pronounced the earlier the therapy was started [63]. In CSF of an adult SMA cohort, levels of pNF-H in CSF were below detection limit, but levels of NSE and pTAU-protein showed a significant decrease under treatment [64]. Electrophysiological biomarkers include the examination of the compound muscle action potential (CMAP) and the motor unit number estimation (MUNE), which have already been used in clinical trials [42]. Availability of validated biomarkers would ideally allow predicting the clinical course of disease and the response to any drug treatment. This would improve clinical decision-making and significantly reduce the time and resources for clinical drug development.
EMERGING NEWBORN SCREENING
A consistent finding across clinical trials for both SMN2 splicing modification and gene therapy is the fact that the effect size depends on the age at treatment initiation: the earlier treatment is started, the greater the clinical benefit is. The most impressive results have been observed when treatment is initiated before the first clinical symptoms become apparent. As we know that denervation progresses rapidly during the first 6 months of life [65], the ‘rescue’ of these motoneurons before clinical deterioration appears to be essential. Nevertheless, the mean age of diagnosis in SMA type 1 is around 6 months of age [66]. Newborn screening (NBS) thus enables us to identify these patients at a pre-symptomatic stage. Four pilot projects of NBS programs in SMA have been conducted and published so far, all using quantitative polymerase chain reaction (qPCR) assays detecting homozygous deletions in either exon 7 [67–69] or intron 7 [70] of SMN1 via dried blood spot analysis. Only one of these assays was validated as also detecting heterozygous carrier deletions [67], and none of the assays was able to detect point mutations or quantify SMN2 copy numbers. In the NBS pilot studies in Taiwan, New York State and Germany, verification of NBS results by sequencing yielded a positive predictive value of 100% [67, 70]. To lower the costs of analysis, different PCR-based assays have been developed that allow simultaneous screening for SMA (with or without SMN2 copy number quantification) and severe combined immunodeficiency (SCID) [71, 72]. SMA was added to the Recommended Uniform Screening Panel (RUSP) in July 2018; NBS for SMA is being implemented in a few US states and southern Belgium, and pilot screening projects are ongoing in other states and countries. Nevertheless, the issue regarding who should be treated is highly controversial [73]. The correlation between SMN2 copy numbers and disease severity was recently examined in a larger Spanish cohort of 625 patients with SMA of all subtypes [17]. Two SMN2 copies were associated with SMA type1 in almost 90% of patients. In patients presenting three and more copies, the individual age of onset and severity are more difficult to predict, but those factors still correlate with the copy number. An algorithm for treatment decisions for children diagnosed with SMA by NBS has been proposed by the SMA NBS Multidisciplinary Working Group, supported by CureSMA. There was consensus among the experts participating in this delphi-technique-based process, namely that treatment should be initiated immediately in truly asymptomatic infants with one SMN2 copy and in infants with two or three copies with or without symptoms, while those with four or more copies should be closely monitored and only treated after the onset of signs or symptoms [74]. However, this pragmatic proposition does not incorporate the presence of possible genetic modifiers in SMA other than the number of SMN2 copies that can mitigate or exacerbate the clinical course [75]. This is also reflected in the observation that disease severity can differ even in siblings possessing the same SMA genotype. The fact that parents of an apparently healthy baby are confronted by a severe diagnosis and difficult treatment decisions furthers adds to the complexity of NBS programs. To address these problems, greater awareness for SMA in the public and the availability of qualified genetic counseling are necessary to help parents make an informed decision [76, 77].
NEW PHENOTYPES, NEW CHALLENGES
Since the introduction of new drug treatments for SMA, we have observed disease trajectories that differ significantly from the known natural history of the disease. These new phenotypes now also cross the traditional subtypes of SMA. For example, patients with onset before six months of age (typical for SMA type 1) might achieve independent sitting (SMA type 2 by definition) if treatment is initiated early. It is now more appropriate to rely on a combination of age of onset, number of SMN2 copies, and age at start of drug treatment rather than the traditional subtypes to define a clinical phenotype of SMA.
These new disease trajectories also mean we must modify and adapt the clinical approach taken. For example, longer survival without ventilatory support following the initiation of drug treatment needs to be considered when counseling the parents of patients with early-onset types of SMA. On the one end of the spectrum, namely in very severe cases entailing a prenatal onset (SMA type 0), drug treatment is not likely to lead to any relevant improvement in motor function, nor will it prevent the need for permanent ventilation; it might therefore be inadvisable. On the other end, initiating treatment in a presymptomatic patient might result in almost normal motor development.
Additional organ involvement, including occurrence of cardiac defects [78], autonomic dysregulation [79] or abnormal fatty acid metabolism [80, 81] has been reported in SMA. SMN protein is known to be highly expressed prenatally in most organs, so that a significant role in organogenesis has been discussed [82]. Further research is needed to understand if systemic treatment of SMN deficiency is of clinical benefit compared to restricted treatment of the central nervous system.
Clinical trials and real world data
In the context of rare diseases, it is almost inevitable that drug approval will be based on weaker evidence than is the case with drugs for common diseases. This issue concerns the disease spectrums investigated in clinical trials, patient numbers, and observation periods. For example, nusinersen was approved for all types and disease stages of SMA despite the fact that the two randomized controlled trials covered only a small proportion of the total SMA population, namely infants and young children presenting a relatively early disease stage. Several centers published their experiences with nusinsersen treatment of SMA type 1 patients of different age groups in the early-access program (EAP) and delivered similar results [83–88]: Age at the beginning of treatment is the most important factor that determines motor response to treatment. Interestingly, no marked difference in motor response between patients with 2 or 3 copies of SMN2 was observed [84, 87]. In contrast to the clinical trials, patient cohorts were heterogeneous regarding ventilation-dependency, need for nutritional support and age; data on patients up to 19 years of age were analyzed in the Italian cohort [85]. More conflicting than the data on motor response are the findings regarding ventilation and nutritional support: In both the German and French cohorts, a significant proportion of patients exhibiting motor improvement had started permanent ventilation or underwent tracheostomy or the placement of a feeding tube. It is unclear whether this reflects a poor treatment effect on respiratory and bulbar function, or a more proactive approach in the participating centers to provide ventilator support or tube feeding.
A mild improvement in the 6MWT was seen in an adult cohort undergoing nusinersen treatment [64]. Overall, there is very little data available extending beyond the cohort of SMA type 1 patients, and it remains difficult for us to predict ‘which effect can be expected in which subtype and at which stage of the disease’ and ‘how long treatment-effects persist’. Furthermore, there are serious logistical and ethical factors that make additional placebo-controlled trials difficult if not impossible to carry out, now that effective treatment options are available.
Considering the limited evidence on the long-term efficacy and safety of novel drug treatments for SMA and their high costs, it is necessary to systematically collect real-world data to improve the basis for clinical decision-making and reimbursement for patients with SMA. Various international initiatives, including the International SMA Consortium Spinal Muscular Atrophy Patient Registry (iSMAC), the SMArtCARE project and the TREAT-NMD network have now established disease-specific registries or are aiming to collect data from different national registries. These projects share standardized data sets for the longitudinal assessment of SMA patients with or without drug treatment [89–91]. Ideally, these initiatives should involve patient representatives in the governance and follow the IRDiRC-recognized “FAIR Guiding Principles” in order to make the collected data Findable, Accessible, Interoperable and Reusable and thus maximise their utility for research [92].
Need for standardized outcome measures
The application of standardized outcome measures is crucial to harmonize real-world data from different registries and to enable the comparison of results. The ideal group of assessments covers and reflects all SMA subgroups, so that selecting the most suitable outcome measures relies on both the functional levels and age of SMA patients. Requirements should accommodate the developmental state of patients: whereas the CHOP INTEND might be best suited for documenting the functional status of severely affected SMA type 1 patients, some items are inappropriate for adolescent or adult patients. Another important issue is the problem of ceiling effects in certain assessments: children exhibiting improved motor function may attain the maximum score in the CHOP INTEND before the age of 2 years, but the HFMSE requires a certain degree of cooperation and has only been validated for application in children above 2 years of age. The motor domains of the Bayley Scales III might be an option to close this gap. As an example, the current SMArtCARE recommendations for standardized evaluation according to age and disease stage are shown in Table 2.
Recommendations for the evaluation of patients with SMA by the SMArtCARE-project. RULM: revised-upper-limb-module; 6-MWT: six-minute-walking-test
Challenges in clinical care
The conventional disease trajectories of the pre-treatment-era are now often modified by new drug treatments. This involves unprecedented challenges and issues regarding motor and non-motor symptoms. In many aspects, this requires that we reconsider earlier blueprints to enable individualized and the most appropriate decision-making.
Despite the improved survival and motor development of symptomatic patients with early onset SMA, these children also exhibit a higher rate of scoliosis during the first years of live. Greater awareness of this risk, and close monitoring of spinal deformities appear crucial to react early and enable the spine to be stabilized via medical orthoses. As many braces interfere with breathing in the more severely affected patients, choosing the ideal device can be difficult. Surgical interventions entailing ‘growing rod’ systems have been reported to be feasible in children with SMA type 1 as young as 4 to 6 years of age and might be an option for younger children with severe scoliosis [93, 94]. However, further experience in this field is needed to balance the risks and benefits of these interventions. Certain orthopedic devices – such as standing frames – have not been used in most SMA type 1 patients, but they appear promising for the prophylaxis of joint contractures and to allow age-appropriate positioning even in more severely affected patients.
Intrathecal application of drugs like nusinersen can be difficult in patients with severe scoliosis [95, 96]. Fluoroscopy may be necessary for lumbar access in these patients, but that involves high cumulative radiation exposure in potentially lifelong therapy in case of nusinersen [97]. Lumbar puncture is especially challenging in patients who have already undergone spinal fusion; some surgeons suggest creating artificial bone gaps during spinal surgery for later lumbar puncture, but we are still waiting for their long-term data [98]. Alternative routes of application, via intrathecal catheter systems [99] or even via cervical puncture [100] have been suggested, despite the fact that nusinersen has only been approved for application via lumbar puncture.
PERSPECTIVE
For the first time in the history of SMA, new treatments like splicing modification and gene therapy are allowing the clinical course to be substantially modified. Additional therapeutic approaches are currently being taken at advanced stages of clinical development and are likely to expand the spectrum of drug treatment options for SMA. This will add to the complexity of care for patients with SMA. To achieve maximum treatment effects, a timely diagnosis and treatment initiation are particularly important. Standard newborn screening seems to be an appropriate tool to attain this goal, although it remains unclear when treatment should be initiated in patients presenting high numbers of SMN2 copies.
When medications for rare diseases come up for approval, there is often only limited evidence available on its long-term effects and safety, and conducting randomized investigations to deliver such evidence is often impossible. Therefore, the only way to generate additional evidence is to collect and analyze real-world data via high-quality, well-monitored patient registries that attempt to avoid bias, so that they provide meaningful results.
Keeping in mind the recent success of drug treatment in SMA, it is important that we do not disregard individual interdisciplinary clinical management, which remains the backbone of SMA treatment, since many patients are left with a significant disease burden despite drug treatment.
DISCLOSURES
Dr. David C. Schorling, participated in workshops sponsored by Biogen and Roche.
Dr. Astrid Pechmann, received compensation for presentations and training activities from Biogen, received research funding from Biogen.
Prof. Dr. Janbernd Kirschner, received research funding and/or compensation for presentations and consulting services from Avexis, Biogen, Ionis Pharmaceuticals, Novartis, and Roche.