
Abstract
Background:
Neuromuscular disorders (NMDs) are clinically and genetically heterogeneous. Accurate molecular genetic diagnosis can improve clinical management, provides appropriate genetic counseling and testing of relatives, and allows potential therapeutic trials.
Objective:
To establish the clinical utility of panel-based whole exome sequencing (WES) in NMDs in a population with children and adults with various neuromuscular symptoms.
Methods:
Clinical exome sequencing, followed by diagnostic interpretation of variants in genes associated with NMDs, was performed in a cohort of 396 patients suspected of having a genetic cause with a variable age of onset, neuromuscular phenotype, and inheritance pattern. Many had previously undergone targeted gene testing without results.
Results:
Disease-causing variants were identified in 75/396 patients (19%), with variants in the three COL6-genes (COL6A1, COL6A2 and COL6A3) as the most common cause of the identified muscle disorder, followed by variants in the RYR1 gene. Together, these four genes account for almost 25% of cases in whom a definite genetic cause was identified. Furthermore, likely pathogenic variants and/or variants of uncertain significance were identified in 95 of the patients (24%), in whom functional and/or segregation analysis should be used to confirm or reject the pathogenicity. In 18% of the cases with a disease-causing variant of which we received additional clinical information, we identified a genetic cause in genes of which the associated phenotypes did not match that of the patients. Hence, the advantage of panel-based WES is its unbiased approach.
Conclusion:
Whole exome sequencing, followed by filtering for NMD genes, offers an unbiased approach for the genetic diagnostics of NMD patients. This approach could be used as a first-tier test in neuromuscular disorders with a high suspicion of a genetic cause. With uncertain results, functional testing and segregation analysis are needed to complete the evidence.
INTRODUCTION
The clinical application of whole exome sequencing has greatly improved the availability of genetic testing and significantly reduced the costs and duration of the diagnostic process for neuromuscular disorders [1–8]. Broad sequencing panels have proven to be useful for these analyses because of the high degree of clinical variation, genetic heterogeneity and pleiotropy, such as in limb girdle muscular dystrophies (LGMDs) and congenital myopathies [7]. Rather than being the final step in the diagnostic process, early application of whole exome sequencing (WES) has been proven to result in a high diagnostic yield and is gradually becoming common practice, both for neuromuscular disorders and for neurological disorders in general [5, 10]. Overall success rates of WES for LGMDs in non-consanguineous populations are around 40–69% [1, 6–8], and even higher (76%) in a population with a high degree of consanguinity [5]. Overall, the putative causative (likely) pathogenic variants were mostly in LGMD-associated genes, but also in genes not included in the current LGMD classification (such as FLNC and DMD) [2, 8]. Finally, the combination of WES with copy number variation analysis enables simultaneous detection of larger duplications and deletions.
Here, we present the results of clinical exome sequencing with bioinformatic panel-based variant filtering for 396 patients suspected of having a genetic neuromuscular disorder with a variable age of onset, neuromuscular phenotype, and inheritance pattern. All patients lacked a definite clinical or molecular diagnosis despite previous ancillary tests (including multiple targeted gene tests in many of them). Our goal was to establish the clinical utility of panel-based WES in neuromuscular disorders (NMDs) in an unselected population of children and adults with neuromuscular symptoms and signs.
In contrast to previous studies on WES in specific cohorts, mainly LGMDs, in a research setting, this is a retrospective observational study in a diagnostic setting in a large unselected group of patients with neuromuscular disorders. As such, this cohort adequately represents the current clinical practice in our reference center for neuromuscular disease.
METHODS
Patients
From 2013 to 2015, DNA of 396 patients with suspicion of a genetic NMD from 16 different countries was sent by their physician ((pediatric) neurologist or clinical geneticist) to the Human Genetics department of the Radboud university medical center for clinical exome sequencing and analysis of a NMD-associated gene panel. Indications mentioned in the order forms were findings suggestive of a genetic neuromuscular disorder on medical history and/or physical examination such as muscle weakness, atrophy, or other muscular signs on physical examination, positive family history for neuromuscular disorder, and abnormal results from ancillary tests such a muscle histopathology or other laboratory findings. All referred patients were included in this retrospective analysis, independent of age of onset, clinical symptoms, or inheritance pattern. As the age at diagnosis was not available for each patient, age at the end of the inclusion period was used.
The cohort consisted of patients in whom a cause for neuromuscular disorders was being sought, both new referrals and revisiting patients. About 53% of the patients was male (211/396; 53.3%). The median age at the end of the inclusion period was 33 years [range 0–81]) (Supplementary Table 1, 2 and 3).
Exome sequencing and data analysis
The Human Genetics department at the Radboudumc has increasingly performed WES, to approximately 4000 patients per year to date, of which approximately 350 have a suspected neuromuscular disorder. In the period of 2013–2015, when exome sequencing was not as common as it is today, the total number of patients with NMD was 396. The clinical geneticist, neuromuscular neurologist and clinical molecular geneticist meet monthly to discuss the results of the panel sequencing. This study adhered to the tenets of the Declaration of Helsinki.
Exome sequencing was performed as previously described [11, 12]. In brief, an Agilent SureSelect Human All Exon 50Mb kit (Santa Clara, CA, USA) was used to capture and enrich exons. Sequencing was performed using an Illumina HiSeq 2000 (San Diego, CA, USA) machine. Read mapping and variant calling were done using BWA and GATK, respectively [12].
A bioinformatic filter for a ‘muscle disorders’ gene panel was applied. This panel consisted of approximately 120–150 genes implicated in various forms of myopathies, muscular dystrophies, myotonic syndromes, and myasthenic syndrome (Versions: DG 2.4x, DG 2.3x, DGD141114, DGD200614, DGD181213 to be found at https://www.radboudumc.nl/en/patientenzorg/onderzoeken/exome-sequencing-diagnostics/exomepanelspreviousversions/muscle-disorders) and is largely based on categories 1–9, 11, 12 and 16 of the GeneTable of Neuromuscular Disorders (http://www.musclegenetable.fr/). Only genes with substantial evidence for causality (multiple families, functional evidence, and/or literature reports) were included in this panel. Genes that cause disease by repeat expansion or contraction as molecular mechanism only were not included. This panel is updated ~3 times per year: the current version (September 2018) consists of > 150 genes. Clinicians can request a reanalysis based on the updated panel.
The detected variants were prioritized based on the following criteria: frequency in the population (<5% dbSNP,<5% Exome Aggregation Consortium (ExAC) database of > 60000 exomes,<1% in-house database of > 5000 exomes), nucleotide and amino acid conservation (based on alignments), relation of the gene to disease (per family), and inheritance pattern [11].
Copy number variant (CNV) analyses from the WES data were done as described [3]. Essentially, it was done using CoNIFER (http://conifer.sourceforge.net/) on the genes in the panel [13]. CNVs affecting genes from the muscle disease gene panel were filtered and only those with an absolute Z-score greater than –1.7 were considered for analysis. To reduce false calls due to potential batch effects, analyses are performed using the most recent samples as controls. CNVs were annotated based on the number of RefSeq exons affected, frequency of CNVs within the cohort, and overlap with disease genes from the muscle disease panel.
Assessment of pathogenicity of variants was performed using the standards and guidelines for the interpretation of sequence variants of The American College of Medical Genetics and Genomics (ACMG) [14].
mRNA analysis
In order to validate the functional effect of the NEB variant c.9946 C>G (r.(spl?) / p.(Arg3316Gly)), mRNA analysis was performed. Fresh muscle tissue was taken from the vastus lateralis muscle and frozen in liquid nitrogen. Muscle was sonicificated to lyse the tissue. Reverse-transcriptase-PCR was done using iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA, USA) and the primers 5’AGTGAGAGCCTCTATCGTCA3’ – 5’TGTATCAGGCATGACATGAATAGT3’. PCR products were sequenced using standard Sanger sequencing.
Clinical information
Once a pathogenic variant was identified, the referring clinician was invited to provide more extensive clinical information on age at onset and phenotype of the patients. Specific questions were on severity and localization of muscle weakness (proximal, distal, axial, facial), ptosis / ophthalmoplegia, spine abnormalities, joints contractures or joint hypermobility, respiratory function, or any additional features, results of muscle biopsy and muscle imaging. We have not requested additional clinical information of the patients with a likely pathogenic variant or variant of uncertain significance, and those with a likely benign variant. No additional clinical information of patients was requested in case of possible pathogenic variants or likely benign variants, because this was not feasible within the time frame of this study.
Statistical analysis
A Mann-Whitney test was used to determine the difference in age at the end of the inclusion period between patients with a definitive genetic cause, likely pathogenic cause, and no likely genetic cause. This was performed to detect whether age of onset is a clinical marker for the chance of detecting a genetic cause.
RESULTS
Exome sequencing
Gene panel analysis of the NMD-associated genes identified pathogenic variant(s) in 19% (75/396) of the patients. In total, 121 pathogenic variants across 46 genes were identified. Pathogenic variants in the three COL6-genes (COL6A1, COL6A2, and COL6A3; 12/75) were the most common, followed by pathogenic variants in the RYR1 gene (7/75) (Figs. 1 and 2; Supplementary Table 1). Together, these four genes account for almost 25% of cases in whom a definite genetic cause was identified. Other relatively frequently mutated genes were ANO5, LMNA, SCN4A, and SELENON, detected in three patients each.
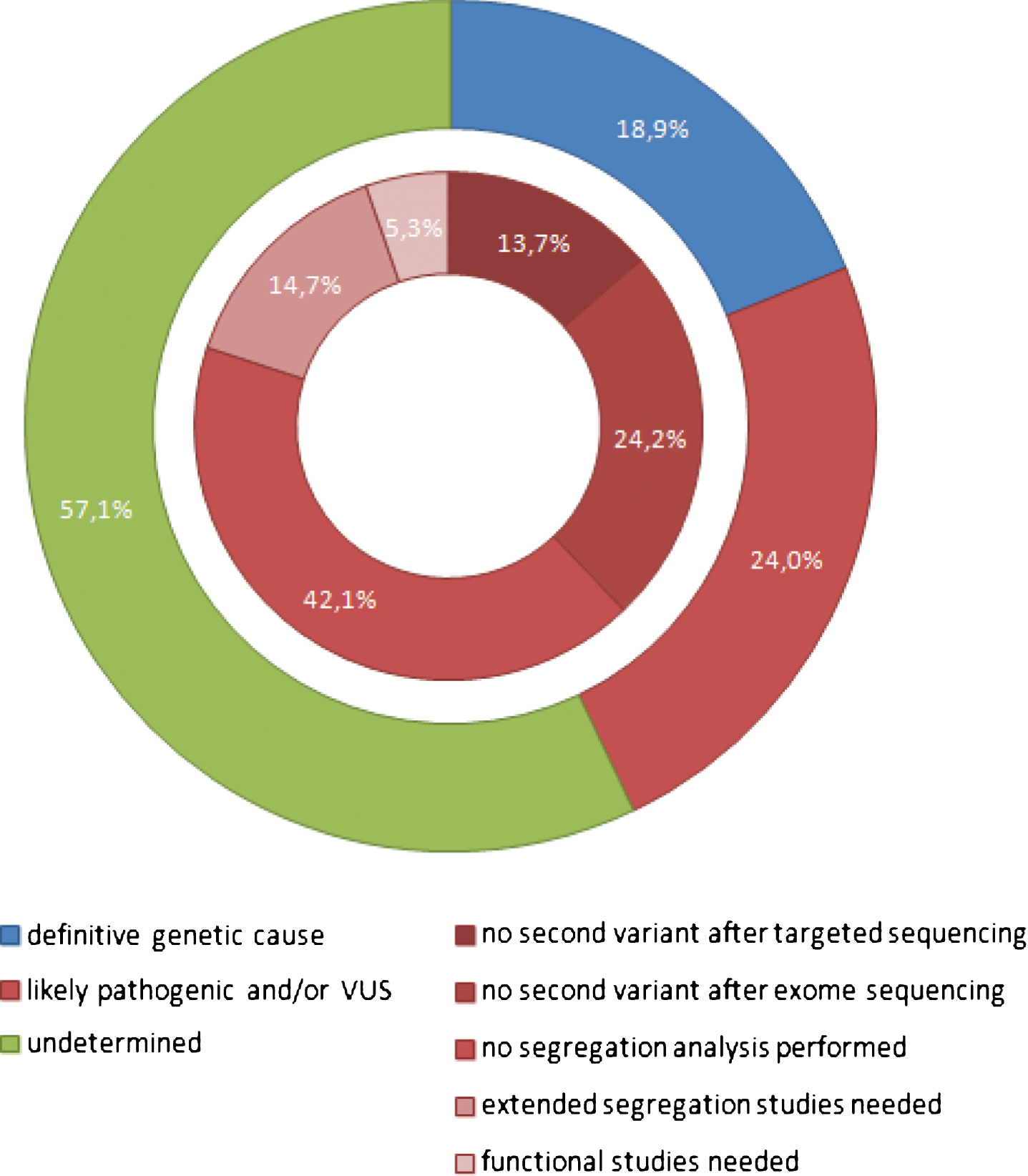
Diagnostic yield and variant distribution of panel based WES analysis in 396 patients. Number of NMD patients are categorized according to findings in clinical exome sequencing after gene panel analysis. The outer circle depicts variant classification based on pathogenicity (pathogenic variant, likely pathogenic variant and/or VUS, undetermined). The inner circle shows the reason why a likely pathogenic variant or VUS could not be classified as pathogenic. VUS indicates ‘variant of uncertain significance’.
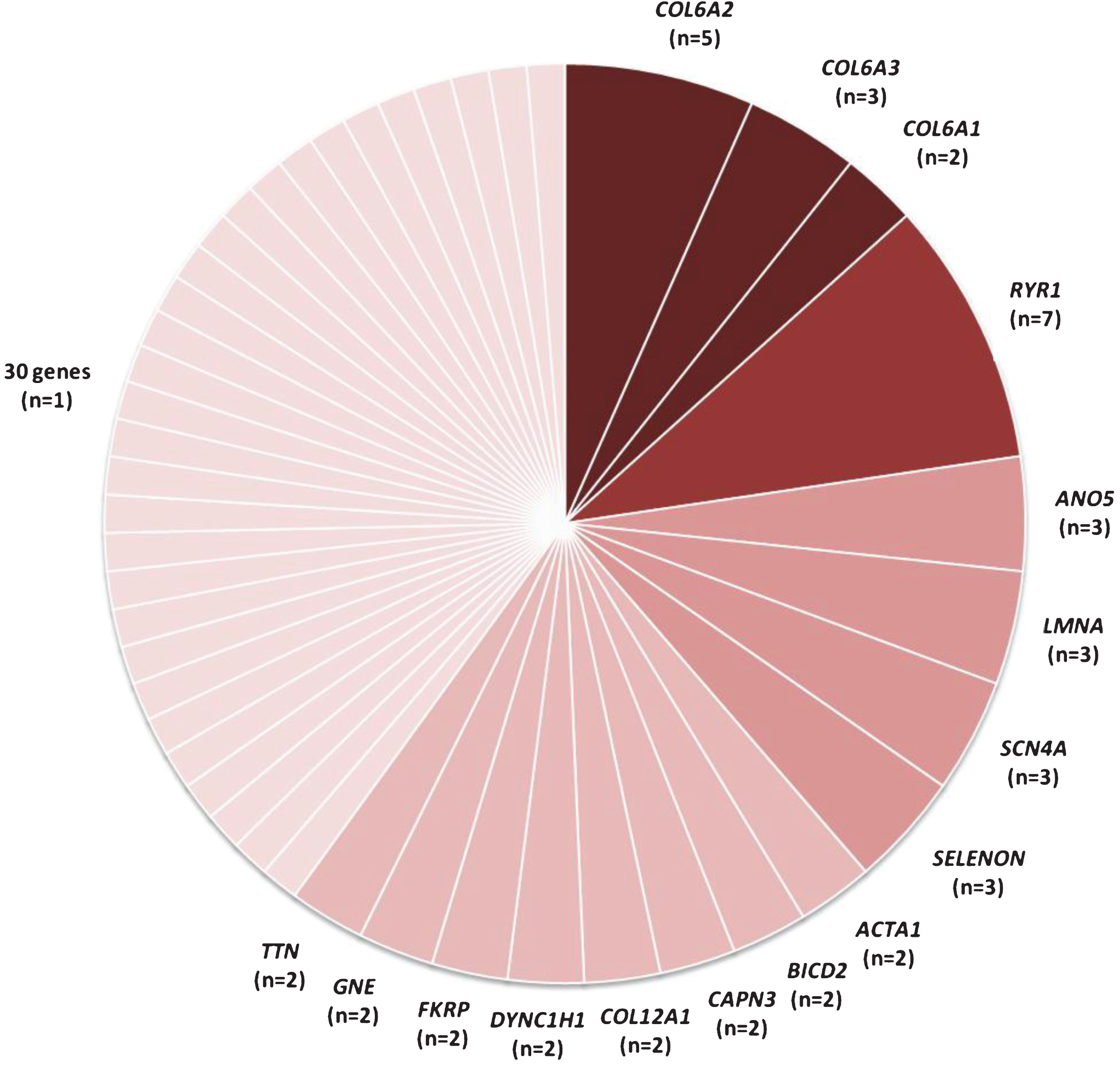
Genes with definitive genetic causes within the neuromuscular disorder gene panel analysis in 396 patients. In 75 patients a definitive genetic cause was detected. The COL6-genes (COL6A1, COL6A2, and COL6A3) combined and RYR1 carried most pathogenic variants and together these genes are responsible for the NMD in almost 25% of the cases (19/75) in whom a genetic cause was identified. Other commonly identified genes were ANO5, LMNA, SCN4A, and SELENON, all with three patients. Genes with variants in only one patient: ATP2A1, CAV3, CHKB, CHRNE, CRYAB, DES, DOK7, ECEL1, EFEMP2, FLNC, GAA, GBE1, HSPG2, IFIH1, KBTBD13, MEGF10, NEB, PLEC, POMT1, POMT2, PYGM, RAPSN, RBCK1, SGCA, SGCG, SLC52A3, TNNT1, TPM2, VCP, and ZC4H2.
An autosomal dominant inheritance was seen in 28 of 75 patients. In 26 patients, segregation analysis was performed: 17 variants were proven to be de novo and the remaining variants were present in affected family members. An autosomal recessive NMD (compound heterozygous or homozygous) was identified in 44 patients, and X-linked inheritance was seen in one patient, in whose healthy mother the variant was present in mosaic form (10%). In two patients, inheritance patterns remained uncertain since a heterozygous pathogenic variant was identified in a gene that can cause both autosomal dominant and autosomal recessive NMD (COL6A2 and CRYAB) while segregation analysis could not be performed.
In 95 of 396 patients (24%), a likely pathogenic variant and/or variant of uncertain significance (VUS) was identified (Figure 1; Supplementary Table 2). In the remaining 226 patients (57%), no variants were found in one of the examined genes (Figure 1).
There were several reasons why likely pathogenic variants or variants with uncertain significance could not to be classified as causative:
CNV analysis in WES data of the whole cohort resulted in the detection of a pathogenic deletion in only one patient (patient 138, Supplementary Table 2). A heterozygous deletion of exons 273–307 within the TTN gene (NM_1333378.4) was identified, in addition to a heterozygous missense variant in the same gene (c.34625T>C (p.(Val11542Ala))). As the missense variant has not been identified before in other patients and no functional information is available, this missense variant has so far been classified as a variant of uncertain significance. Therefore, a limb-girdle muscular dystrophy due to two TTN variants could not be confirmed, though a risk of a dominant cardiomyopathy (with incomplete penetrance) because of the large TTN deletion is likely.
Clinical information
We received additional clinical information on 55/75 patients with a definitive genetic cause (response rate of 73%) (Table 1). Age at onset was≤1 year in 27 patients, between 1 and 18 years in 16 patients,>18 years in 10 patients and unknown in two patients. Overall, the mean age at the end of the inclusion period of the patients with a definitive genetic cause was 25 years (range 0–66). As such, this group was younger than the groups of patients with a likely pathogenic variant and/or VUS (mean age 34 years, range 0.8–80.8; p = 0.0061) and of patients without a genetic cause (mean age 36 years, range 0.6–77.1; p = 0.0002). This is consistent since severe phenotypes are more likely to be caused by an obvious genetic aberration, and the chance of having an acquired cause of the neuromuscular disorder is smaller in younger patients.
Summary of diagnoses and clinical findings in 55 of 75 patients with a definite genetic diagnosis due to a pathogenic variant, of whom we received more detailed clinical findings. Clinical phenotype, muscle biopsy findings, results of EMG, muscle-MRI and ultrasound, and laboratory findings are shown
1Identified genetic variants are shown in Supplementary Table 1. 2To our knowledge, three patients had passed away before NGS was requested (P2, P46 and P54; indicated with † at the specific patient numbers). 3Normal levels CK [21].
In 15/55 (27%) of patients in whom a definitive genetic cause was identified, whole exome sequencing was the first genetic test performed. In the other patients (40/55; 73%) at least one genetic test was performed before (karyotyping, SNP array, hotspot gene testing, testing of one to 15 genes), as shown in Table 1. For example, in the patients with a definitive causative COL6A1/2/3 variant, previous tests were Sanger sequencing of ACTA1, RYR1, SELENON, CFL2, TMP2, TMP3 (in patients with a congenital myopathy with onset≤1 year and increased fiber size variability on the biopsy). The phenotypes of myopathy without typical long finger flexor contractures had apparently not pointed to a collagen-6 myopathy. In a patient with adult onset limb-girdle weakness, ANO5 and CAPN3 genes were tested, and a patient with predominantly shoulder girdle muscle weakness was tested for fascioscapulohumeral dystrophy type 1. In five of seven patients with a variant in RYR1 other genetic tests had been performed. This might be related to the muscle biopsies which had shown abnormalities compatible with a congenital myopathy (such as increase in internal nuclei, increase in fiber size variation, or congenital fiber disproportion but no cores). None of the patients with the genetic diagnosis of congenital myasthenic syndrome (CMS) (with pathogenic variants in CHRNE, DOK7, and RAPSN; patient 13, 28, and 52 respectively) were recognized clinically as CMS, but as having atypical forms of FSHD, LGMD, or a mitochondrial disease (Table 2). In Table 2, other potential reasons for the delay in diagnosis are shown (i.e. new genes or diseases, unusual clinical features, biopsy results indicating towards an acquired NMD).
Potential reasons for diagnostic delay in 10 selective patients eventually receiving a definitive genetic diagnosis
Follow-up analyses
Exome sequencing sometimes generates hypotheses, rather than confirming clinical diagnoses. As a consequence, proper follow-up should be considered. This may consist of reverse phenotyping, functional studies, pathology and / or segregation analyses. Below we present three cases as examples where such studies were conclusive. The first case emphasizes that actual clinical assessment might be needed to complement genetic segregation analysis in disorders of variable clinical severity, to prevent unjustified disregard of a variant found in an apparently asymptomatic person. The second case shows that WES analysis can reveal an unexpected but treatable condition, that retrospectively explains many health problems experienced for a very long period. The patient was classified as having an atypical form of FSHD or LGMD at the time of presentation. Only later, the history of ‘having difficulties walking in childhood’ was considered to be an early manifestation of the congenital myasthenic syndrome. This group of disorders was barely known at presentation and a neuromuscular junction disorder had not been considered. Repetitive nerve stimulation was therefore only performed after the results of panel-based WES and confirmed the presence of a neuromuscular disorder. The third case illustrates the importance of functional analysis to confirm pathogenicity. The results of mRNA analysis enabled the clinical geneticist to counsel this family in prenatal testing options in future pregnancies.
CASE 1: PATIENT 24
This patient presented at the age of 18 with muscle weakness in a limb girdle distribution since the age of 15 with a history of neonatal hip dysplasia. He reported myalgia between his shoulders and in his proximal arms and legs. He already had difficulties with running in high school. He had mild proximal weakness of his arms (manual muscle testing according to the Medical Research Council grading (MRC): 4 of rhomboid, biceps and triceps muscles), and mild contractures of Achilles tendon and elbow were observed. His skin showed hyperkeratosis pilaris (Figure 3). He reported no family members with similar symptoms.
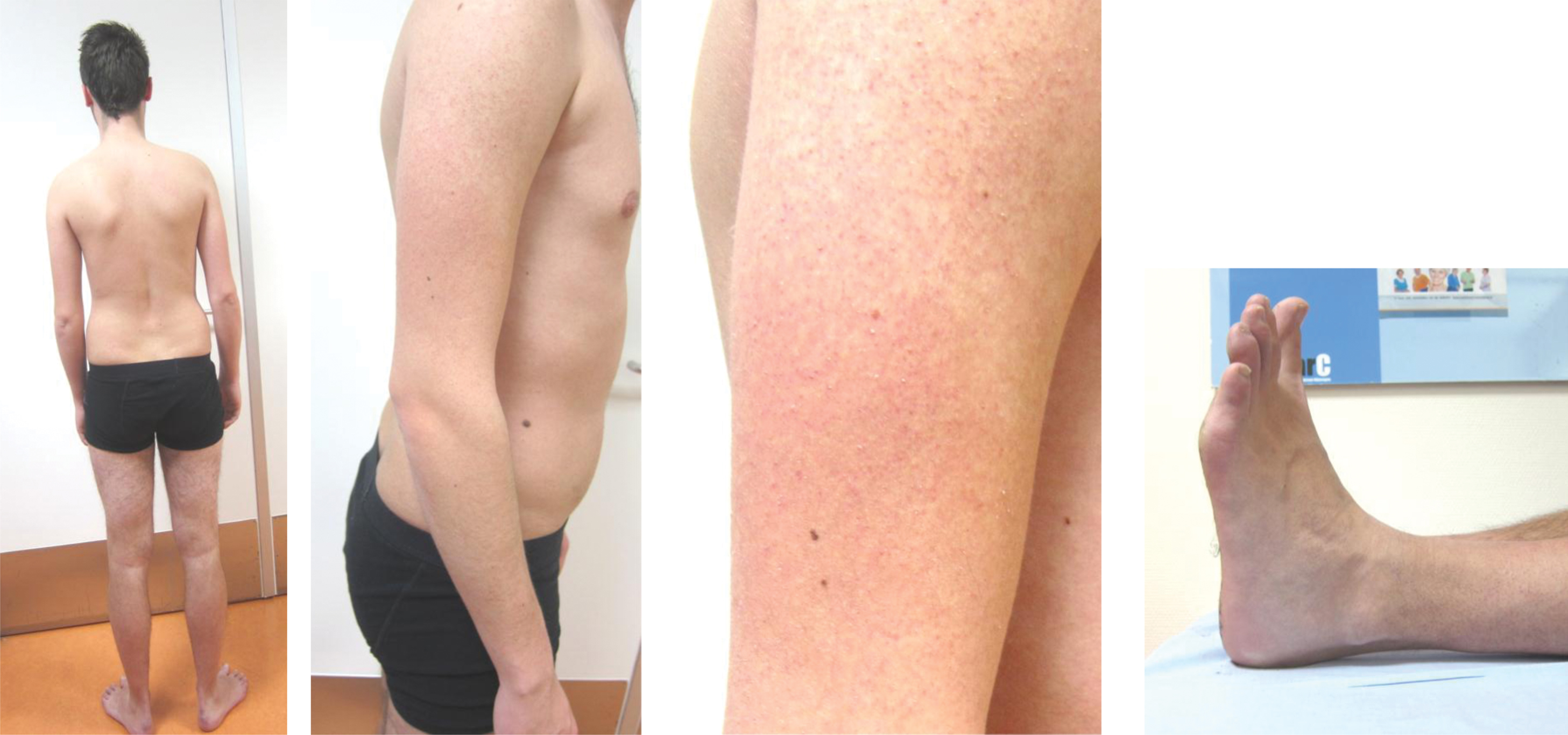
Clinical features of Case 1: patient 24. This 18-year old patient presented with limb girdle muscle weakness with neonatal hip dysplasia since the age of 15. Examination showed mild proximal weakness of his arms (MRC4 rhomboid, biceps and triceps), with mild winging of left scapula. Mild contractures of Achilles tendon and elbow. His skin showed hyperkeratosis pilaris. WES showed a heterozygous variant in COL6A3 (c.6130 G>A (p.(Gly2044Arg)), which was also found in his mother who was initially considered to be unaffected but later showed very similar features on history and examination. Printed with permission of patient.
The CK was elevated (829 U/l), and needle electromyography showed small polyphasic motor unit action potentials. FSHD1 had been excluded by genetic testing. A muscle biopsy (vastus lateralis muscle) showed mild increase of fiber size variation. Panel-based WES analysis showed a heterozygous variant in COL6A3 (c.6130 G>A (p.(Gly2044Arg)), which was considered to be pathogenic since it involves a glycine change in the triple-helix domain. Furthermore, this variant has been reported before in a patient with a congenital myopathy [15].
Segregation analysis in his seemingly unaffected mother showed the same variant. She reported having had difficulties in sports at school. She had a scoliosis for which she had needed a brace in her teenage years. On physical examination she had mild shoulder girdle weakness (MRC 4 deltoid, rhomboid, biceps and triceps) and also keratosis pilaris of the skin. A muscle ultrasound showed increased intensity in most muscles scanned, compatible with a neuromuscular disorder.
The conclusion was that both the patient and his mother were affected by the same neuromuscular disorder (LGMD D5 collagen-6 related / Bethlem myopathy) caused by the pathogenic variant in COL6A3. The mother had never considered herself to be affected, despite limitations in motor functioning throughout her life. Nevertheless, she was very pleased with the results of the genetic test and neurological investigation, since it explained many symptoms retrospectively, and encouraged her to accept support and training advices. Genetic counseling was performed, and both the patient and this mother were referred to a rehabilitation center.
CASE 2: PATIENT 28
This 62-year old patient complained of increasing motor symptoms. He had been clinically diagnosed before as atypical FSHD with a myopathy with predominantly axial and shoulder girdle weakness. Previous molecular tests had excluded FSHD1 and 2, Emery–Dreifuss muscular dystrophy, and LGMD R1 calpain3-related. He reported gradually progressive limitations in walking. His walking distance was limited to 1.5 hours, whereas he used to be able to walk for six hours. Walking upstairs had become more difficult. Furthermore, his voice had become softer, he had difficulties swallowing, and he could no longer raise his arms above shoulder height. Retrospectively, he reported having had an episode of severe muscle weakness of his legs, which caused an inability to walk for weeks, at the age of 10. He had noticed his first shoulder girdle symptoms when he started working as a teacher at the age of 21. His older brother had had similar problems and was diagnosed with FSHD; he died at the age of 64 due to myocardial infarction. One sister had died in early childhood, without a known cause. Physical examination showed atrophy of the shoulder girdle muscles and of proximal arms, with hypertrophy of the trapezius muscle. Active shoulder abduction was limited to 90 degrees, and anteflexion to 110 degrees. There was mild scapular winging. Neck flexion and extension (MRC 3), and elbow flexion (MRC 4) were weak (Figure 4).
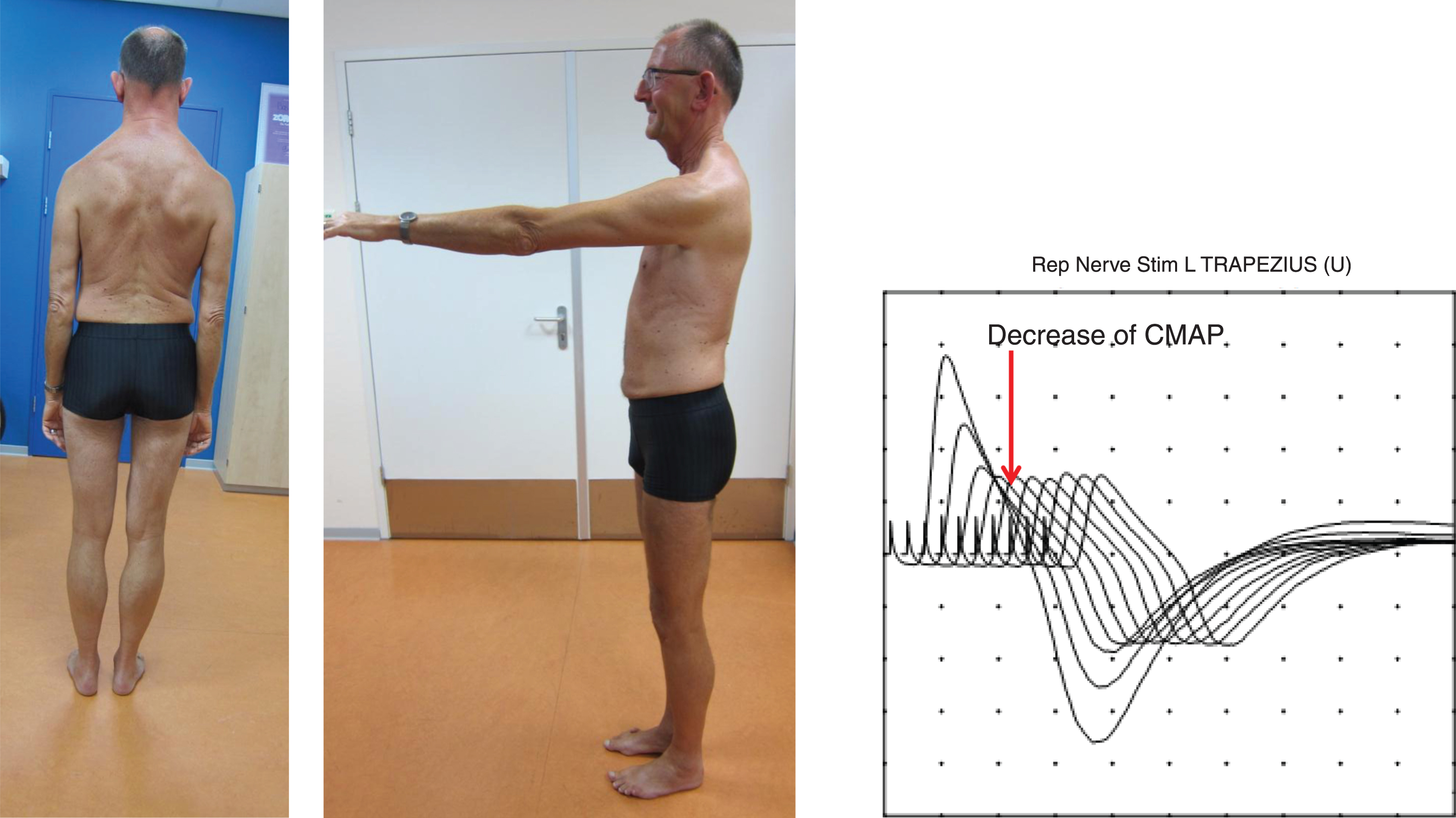
Clinical features of Case 2: patient 28. Physical examination showed atrophy of shoulder girdle muscles and of proximal arms, with hypertrophy of the trapezius muscle. Shoulder abduction was limited to 90 degrees, and anteflexion to 110 degrees. There was mild scapular winging. Neck flexion and extension (MRC 3), and elbow flexion (MRC 4) were weak. Repetitive nerve stimulation (3 Hz) of accessory nerve showed 57% decrement (decrease of the compound muscle action potential (CMAP) measured by surface electromyography) of the trapezius muscle between the 1st and 4th stimulus. Printed with permission of patient.
Needle electromyography showed small polyphasic motor unit action potentials in proximal arm muscles. A muscle biopsy (vastus lateralis muscle) at the age of 54 had shown increased fiber size variation, but no other abnormalities on enzyme and immunohistochemical staining. MRI of the muscles (age 62) showed mild fatty infiltration of the axial muscles but no other abnormalities. Panel-based WES analysis showed a homozygous pathogenic variant in DOK7, c.1124_1127dup (p.(Ala378fs)), causing myasthenic syndrome type 10. His parents were not known to be consanguineous. Segregation analysis in the affected brother and sister was not possible due to lack of material. Subsequent repetitive nerve stimulation showed decrement after the second stimulus, which further decreased in the 2nd to 5th response, matching with myasthenic syndrome. Treatment with salbutamol 2 mg three times a day significantly increased his exercise tolerance and voice volume, and slightly improved his muscle strength.
CASE 3: PATIENT 47
This 5-year old female patient presented at birth with severe hypotonia. She was admitted at the intensive care and was not able to swallow, for which tube feeding was started and continued. Non-invasive ventilation was started at the age of 2 years. At the age of 5, physical examination revealed an open mouth, facial weakness, and scoliosis. Muscle weakness was very pronounced, proximally MRC 2 and distally 0 to 3. She used an electric wheelchair and was ventilated approximately 80% of the day.
A muscle biopsy at 8 months of age had shown central cores and multiminicores. Electron microscopy also revealed rods. Subsequently, Sanger sequencing of SEPN1 and RYR1 had been performed and no pathogenic variants were detected. Four years later, panel-based WES showed two heterozygous variants in the NEB gene, a maternal pathogenic frameshift variant (c.18745_18748dup; p.(Asn6250fs)) and a paternal variant (c.9946 C>G; r.(spl?) / p.(Arg3316Gly)), that was predicted to enhance a cryptic donor splice site in exon 70 (Figure 5A). A fresh frozen muscle biopsy from the healthy father was used for targeted mRNA studies. This indeed showed that the cryptic splice donor site was used in the mutant transcript only (Figure 5B), while this cryptic site was not used in a control sample. The use of this donor splice site results in a transcript that lacks 197 nucleotides (out of frame). The signal of this aberrant transcript is weaker (lower peaks in sequencing reads), likely due to nonsense-mediated decay (not tested).
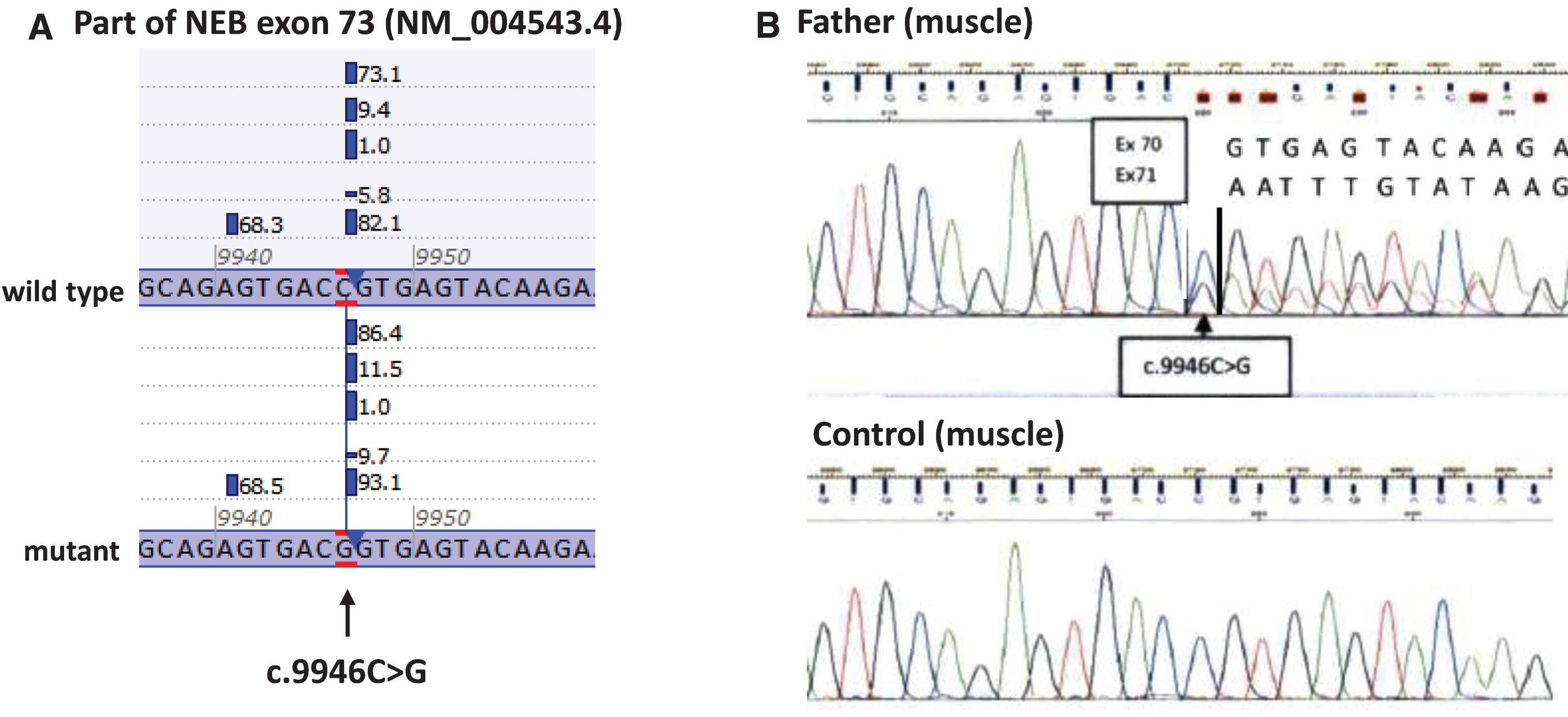
Results of mRNA analysis of the c.9946 C>G (p.(Arg3316Gly)) variant in NEB on fresh frozen muscle biopsy of the healthy carrier father of patient 47 (Case 3). As shown in Figure 5A, splice prediction software predicted an enhancement of a cryptic splice donor site in exon 73 (12–50% increase). This cryptic splice donor site is used in the muscle of the father, leading to 30–40% (Figure 5B) of the transcript, lacking 197 nucleotides. This mutant transcript leads to a frameshift and most probably to nonsense-mediated decay (hence the 30–40% mutant transcript).
DISCUSSION
This retrospective observational study in a large unselected group of patients with presumed neuromuscular disorders resulted in a definite genetic diagnosis for 19% of patients. In this group, pathogenic variants in the three COL6-genes (COL6A1, COL6A2, and COL6A3) were the most common cause of the identified muscle disorder, followed by RYR1. Together, these four genes were causative in almost 25% of cases in whom a genetic cause was identified. A likely pathogenic variant and/or variant of uncertain significance was detected in 24% of patients, and in 57% of patients no genetic aberration was found that might explain the neuromuscular disease.
Overall, our results show that panel-based WES seems an effective manner to identify molecular causes in neuromuscular disorders, both as subsequent test as well as a first-tier test. The advantages of WES are its unbiased approach, detecting variants in genes that would not be tested based on the clinical phenotype (Table 2), and the fact that it offers new diagnostic options in patients in whom Sanger sequencing has so far not resulted in a specific genetic diagnosis. It may also be cost-effective to do WES as first-tier test unless there is high certainty of a single gene cause, such as in spinal muscular atrophy (SMN) or in malignant hyperthermia (RYR1 in the large majority of patients). This accounts for the current diagnostic setting in the Netherlands, where neuromuscular WES (filter or panel) is available in a number of academic centres, is paid for by the health insurance, is less expensive than testing two subsequent genes with Sanger sequencing, and takes the same time as Sanger sequencing of one gene. This varies in different countries based on price and availability of WES and local legal policies.
The diagnostic yield of 19% seems low compared to series published recently (30–76% in cohorts of patients with limb-girdle muscular dystrophy with myopathic or dystrophic changes in the muscle biopsy, some of which were populations with a high degree of consanguinity [2, 8]). There are several reasons to explain this. First, we included all patients which were sent for WES analysis to our department. This implied both patients with a high and low suspicion of a genetic neuromuscular disorder, resulting in a very heterogeneous cohort. Some of them may possibly have an acquired rather than a genetic cause of their neuromuscular symptoms, such as a anti-HMGCR myopathy [16]. Second, our cohort includes DNA of both new referrals and of revisiting patients. Third, follow-up studies (segregation analysis or functional testing) has revealed that many variants of uncertain significance in these previous studies were in fact not pathogenic, reducing the ‘diagnostic yield’. Hence, we consider the percentage of 19% definite and 24% possible genetic diagnoses quite high given the fact that our strategy is the use of panel-based exome sequencing in case of any suspicion of an inherited neuromuscular disorder without imposing too many restrictions, as well as a strive to follow-up to determine the pathogenicity of variants. One example is the functional characterization of a SCN4A variant which we were able to perform after WES; this clearly showed the pathogenicity of the mutation (patient 63) [17]. We have not been able to do this for all variants of uncertain significance.
The group of 95 patients with likely pathogenic variants or variants of uncertain significance included 14 patients with variants in more than one gene. In these patients, the pathogenicity of the variants could not be confirmed or rejected due to lack of segregation analysis and additional clinical information. These further interpretation steps might have been performed in the referring center or department without sharing the final conclusions with us (incomplete response rate). The decreasing sequencing costs may prompt us to perform family (trio-based) sequencing whenever possible, to overcome the lack of segregation data that aids in the interpretation of sequence variants.
Panel-based WES in our series as well as those published may have other limitations. In our test, for example, the presence of small CNVs remains undetected since the analysis with Conifer does not allow the detection of deletions smaller than three exons. In this cohort, we have detected only one CNV, a deletion of 34 exons of the TTN gene (patient 139), but smaller deletions exist. Additionally, disease-causing variants outside the coding sequences (i.e. deep intronic, promotor, etc.) of the analyzed genes or variants in genes not yet associated with neuromuscular disorders will not be detected. Finally, WES with a median coverage of 100x is good for germline variants, but has its limitations in the detection of mosaics [18], especially if these mosaics are not present in the tested cell types (lymphocytes). Reanalyses with other CNV detection tools, with updated gene panels, or using the full exome sequencing data set may identify further variants from the same dataset in the future. In the future, whole transcriptome sequencing from muscle biopsies or whole genome sequencing may be helpful to find causative variants in the remaining patients [19].
As certain ancillary investigations, like muscle biopsy and muscle MRI, might lead to ambiguous and inconclusive results, panel-based WES could potentially be the best first-tier test in suspected neuromuscular disorders, which can be complemented by the appropriate functional testing and segregation analysis if needed. For example, in none of the patients with a RYR1 variants, muscle biopsy had shown cores which had directed to RYR1 sequencing in the past. Nevertheless, the clinical phenotypes in these patients were suggestive of a congenital myopathy (perinatal onset or childhood onset, proximal muscle weakness, facial weakness, ophthalmoplegia). In some cases, EMG findings with a high positive and negative predictive value can be used as a functional in vivo confirmation of the genetic results from WES, such as the finding of myotonic discharges in myotonic syndromes or the finding of a CMAP amplitude decrement in a myasthenic syndrome (as illustrated in case 2). Similarly, imaging (MRI or ultrasound) might reveal typical features of collagen-6 myopathies [20]. Immunohistochemistry and Western blot of muscle biopsy are also still valuable diagnostic tools. However, since they test predominantly the presence and amount of protein caused by loss of function mutations, these tests will not detect the variants that cause functional changes of the protein. Hence, as known, WES can be used as an unbiased diagnostic approach for patients with disorders with a high degree genetic heterogeneity such as limb girdle muscular dystrophies and congenital myopathies. This also accounts for collagen-6 myopathies, since the clinical phenotype typical for Bethlem myopathy or Ullrich congenital muscular dystrophy <ins cite="mailto:z900127" datetime="2019-04-17T15 : 27"> (LGMD D5 or R22 collagen-6 related)< /ins>cannot distinguish between variants in COL6A1, COL6A2, or COL6A3. Many of the referrals for neuromuscular WES indeed included a specific description of the phenotype. In some of these patients, Sanger sequencing of one gene could have led to the specific diagnosis; however, it would not have resulted in a diagnosis in others.
The most important limitation of this study is its retrospective design, which is likely to have contributed to an incomplete response to our request for additional information (73%). This limits the amount of clinical information on this group of patients, and the possibilities of determining phenotype – genotype correlations. On the other hand, this reflects the practice in a daily diagnostic setting, in which the clinical data accompanying genetic test applications is often very limited. If only limited amount of clinical information is provided, the interpretation of WES results can be very difficult, for instance in cases with variants in more than one gene (e.g. patient 152 with variants in CHRNA1, COL6A1, FLNC, NEB, and RYR1). Without additional clinical information and segregation data, it is impossible to define which variant is causative. We underscore that WES as a first-tier test in suspected neuromuscular disorders will be a much stronger tool if more complete and precise phenotypical descriptions and feedback are provided and segregation testing is possible.
To conclude, this retrospective observational study in an unselected cohort of 396 patients with presumed neuromuscular disorders resulted in a definite genetic diagnosis in 19%, and in the identification of a likely pathogenic variant and/or variant of uncertain significance in 24% of patients. In contrast to previous studies in strictly selected groups, this study reflects the current application and yield of panel-based WES in a broad diagnostic setting, both for revisiting patients without a genetic diagnosis and for newly manifesting patients. Hence, panel-based WES offers an unbiased approach which could be used as a first-tier diagnostic test in neuromuscular disorders with a high suspicion of a genetic cause and genetic heterogeneity, after which functional confirmation and segregation analysis is sometimes needed to round the interpretation. Clinical reasoning based on history and physical examination, nevertheless, remains the starting point of the diagnostic process and should preferably be performed by physicians with an expertise in neuromuscular disorders.
CONFLICT OF INTEREST
The authors have no conflict of interest to report, and received no financial support for performing this study.