
Abstract
Background:
Congenital myopathies are a group of neuromuscular disorders that typically present at birth or early childhood with hypotonia and non-progressive or slowly progressive muscle weakness. They are classically subclassified by characteristic structural changes and histopathological findings in skeletal muscle. Variants in over 40 genes have been described to date in patients with various forms of congenital myopathy with overlapping phenotypic and histological features, which poses a challenge for laboratories and clinicians in interpreting genetic findings.
Objective:
The purpose of this study was to evaluate the evidence supporting each gene-disease relationship and provide an expert-reviewed classification for the clinical validity of genes involved in congenital myopathies.
Methods:
The ClinGen Neurological Disorders Clinical Domain Working Group assembled the Congenital Myopathies Gene Curation Expert Panel (CongenMyopathy-GCEP), a group of clinicians and geneticists with expertise in congenital myopathies tasked to perform evidence-based curation of 50 gene-disease relationships using the ClinGen semiquantitative framework to assign clinical validity.
Results:
Our curation effort resulted in 35 (70%) Definitive, eight (16%) Moderate, six (12%) Limited, and one (2%) Disputed disease relationship classifications. The summary of each curation is made publicly available on the ClinGen website.
Conclusions:
Expert-reviewed assignment of gene-disease relationships by the CongenMyopathy-GCEP facilitates accurate molecular diagnoses for congenital myopathies and can allow genetic testing to focus on genes with a validated role in disease.
Introduction
With the integration of next-generation sequencing (NGS) in clinical molecular diagnostics, laboratories can test multiple genes that have been reported in association with a disorder on their diagnostic gene panels. 1 Multi-gene panels that allow simultaneous testing of multiple diseases with shared clinical presentations are beneficial in the diagnosis of conditions with phenotypic overlap.2,3 However, NGS panels that include genes with limited or ambiguous role in a disease may lead to uncertain results and variants identified in such genes may be misinterpreted as causative, resulting in misdiagnosis and potentially unnecessary or detrimental therapeutic intervention. 1 Variants in a gene cannot be accurately interpreted if there is uncertainty in the gene's role in a particular disorder.4,5 Therefore, careful assessment and stringent classification of a gene's causative role in disease is a crucial step for correct molecular diagnosis of patients. 5 This is particularly important for diseases that have high genetic heterogeneity with wide phenotypic overlap between implicated genes, as the diagnosis that directs clinical management and genetic counseling of family members relies on the genetic finding.
Congenital myopathies are a group of neuromuscular disorders that typically present at birth with hypotonia and muscle weakness. 6 Patients may also have respiratory insufficiency and feeding difficulties. Cardiac muscle involvement leading to cardiomyopathy and/or arrhythmias is less common but may also be observed in some forms.7–9 As opposed to muscular dystrophies that are characterized by progressive muscle degeneration and wasting, congenital myopathies tend to be nonprogressive or slowly progressive and do not involve continuous degeneration and regeneration in the muscle. Congenital myopathies are classically defined by characteristic structural changes in the skeletal muscle, which can be referred to as the “histotype”, that forms the basis for their clinicopathologic sub-classification, such as nemaline myopathy, centronuclear myopathy, central core disease, multiminicore disease, and congenital fiber type disproportion. 10 Many patients may have a combination of multiple features and overlapping muscle biopsy findings. To date, more than 40 genes have been reported as causal for various forms of congenital myopathies.11,12 A single gene may be associated with more than one clinicopathologic presentation and many genes have been associated with a wide phenotypic as well as histological spectrum, including overlapping features with other related genes.11,12 All this complexity adds challenges to interpreting genetic findings. Establishing the validity of each reported gene's role in congenital myopathies by clinicians and geneticists with expertise in this field would be beneficial to facilitate accurate molecular diagnoses. Towards this goal, the Clinical Genome Resource (ClinGen), 13 a National Institute of Health (NIH)-funded initiative, has assembled a group of congenital myopathy experts to curate disease-implicated genes using a systematic evidence-based framework. 5 Here we present the classification of 50 gene-disease relationships for congenital myopathies by the ClinGen Congenital Myopathies Gene Curation Expert Panel (CongenMyopathy-GCEP).
Materials and methods
Clingen congenital myopathies gene curation expert panel (CongenMyopathy-GCEP)
The CongenMyopathy-GCEP (https://www.clinicalgenome.org/affiliation/40031/) consists of a coordinator, experts with substantial knowledge of congenital myopathies from both clinical and research settings, as well as of biocurators trained in ClinGen methodology to curate evidence and apply the ClinGen gene-disease validity framework in the classification of genes related to congenital myopathies. These roles are filled by an international panel providing diverse experience, including members from the United States, Canada, the United Kingdom, France, Italy, Estonia, and Finland, representing 14 institutions.
Selection of genes and diseases for analysis
In total, 37 genes that have been reported as potentially associated with congenital myopathies were prioritized for curation based on expert knowledge and literature search by the CongenMyopathy-GCEP. Four genes that are often related to dystrophic changes on muscle biopsies but have a predominant congenital myopathy phenotype (COL6A1, COL6A2, COL6A3, and LAMA2) were also included in curation by the CongenMyopathy-GCEP, following consultation with the Limb Girdle Muscular Dystrophy GCEP.
The CongenMyopathy-GCEP implemented standard ClinGen practice for genes with assertions relating to multiple diseases in the Online Mendelian Inheritance in Man (OMIM) database and/or the literature. Following the guidelines from the ClinGen Lumping and Splitting Working Group 14 the CongenMyopathy-GCEP evaluated the mechanism of disease, phenotypic features, and mode of inheritance of each condition to determine whether they should be lumped into one curation record or to split into individual records. Genes that were considered to be associated with a broad spectrum of histological findings (the histotype), rather than specific forms, were linked to disorders based on their histological features. Unique identifiers from the Mondo disease ontology (https://mondo.monarchinitiative.org/) were assigned to each curated disease entity. To include the wide spectrum of phenotypes associated with such genes, their associated diseases were given protein- or gene-based names, such as Actinopathy (for ACTA1-related myopathies) or SELENON-related myopathy.
Clingen gene-disease clinical validity assessment for congenital myopathy genes
The CongenMyopathy-GCEP assessed the clinical validity for 37 genes in relation to congenital myopathies using the ClinGen Gene Curation Standard Operating Procedure, Versions 6–9 (https://clinicalgenome.org/docs/gene-disease-validity-standard-operating-procedure-version-9/) based on the framework described in Strande & Riggs et al.. 5 In short, a structured and evidence-based evaluation of genetic data, such as variants in the gene identified in patients with disease of interest, including case reports, family studies, and case-control studies, and experimental data, including biochemical assays on protein function or expression and animal models available in the literature was performed. A maximum of 12 points and six points were assigned for genetic and experimental evidence, respectively, for a combined maximum total of 18 points. Gene-disease relationships were classified based on the strength of supportive evidence as follows: Definitive (12–18 points, with replication over time), Strong (12–18 points), Moderate (7–11 points), Limited (0.1–6 points), “No known disease relationship” (no reports have directly implicated the gene in human disease cases), Disputed and Refuted (gene-disease relationship has conflicting evidence). To classify a gene-disease relationship as “Definitive” the disease role of the gene with Strong evidence would need to be repeatedly demonstrated in research and clinical diagnostic settings and upheld over time (minimum of three years). The evaluated evidence and scores assigned by the expert group for each gene-disease relationship were recorded in ClinGen's gene curation interface and a summary was published on the ClinGen website (https://search.clinicalgenome.org/kb/affiliate/10031) upon expert approval.
The ClinGen gene curation framework provides guidance, and each expert panel is allowed to adjust scoring or final classifications based on the relevant disease area and the judgment of the expert panel members. For this purpose and to maintain consistency across multiple biocurators, the CongenMyopathy-GCEP developed a guidance document to highlight the salient information for curators to record and consider during scoring (Supplemental Table 1). Genetic evidence scores were reduced when adequate clinical details and previous testing results were not available.
Once the relevant gene-disease relationships were identified, a preliminary evaluation was performed to determine the strategy for curation and review (Supplemental Figure 1). Curations underwent either expedited or standard review processes, following the methods outlined in Ross et al., 2023. 15
Variant classification analysis in ClinVar database
Classification of variants in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/; accessed 10/28/2024) were assessed for each gene. Only single-gene variants that had classifications associated with myopathy-related conditions were included in the analysis; variants that only had classifications related to central or peripheral nervous system disorders or cardiac disorders were not included. For genes that have been classified by the CongenMyopathy-GCEP for more than one congenital myopathy-related condition, the highest level of evidence classification was considered for this analysis.
Results
Thirty-seven genes were curated in association with congenital myopathy disorders. The summary of each curation is made publicly available on the ClinGen website (https://clinicalgenome.org/affiliation/40031/). Each gene was evaluated to determine the most appropriate gene-disease relationship for curation (Table 1). Thirteen of the 37 genes were determined to be related to a broad histotype spectrum of disease that encompassed two or more histological features, including the presence of nemaline rods, cores, centralized nuclei, and/or fiber-type disproportion. Eleven genes (ACTA1, BIN1, COL6A1, COL6A2, COL6A3, MYH2, NEB, RYR1, TNNT1, TPM3, TTN) were considered to be associated with diseases that encompass both autosomal dominant (AD) and autosomal recessive (AR) modes of inheritance. Eight genes were defined in relation to nemaline myopathy, six to centronuclear myopathy, and three to congenital myopathy without notable defining features. Two genes that encode critical components of calcium release-activated calcium (CRAC) channels (STIM1 and ORAI1) were evaluated in association with a combined spectrum of tubular aggregate myopathy with varying severity. Others, such as MYO18B, were associated with a disorder phenotypically distinct from other congenital myopathies, such as fusion of the cervical spine combined with myopathy in Klippel-Feil syndrome. For genes that have been implicated in other disorders not representing a continuum of congenital myopathy phenotypes, such as DNM2 with Charcot-Marie-Tooth Disease, the CongenMyopathy-GCEP focused only on the gene's association with congenital myopathies (Table 1).
Establishment of gene-disease relationship and mode of inheritance for each curation record.
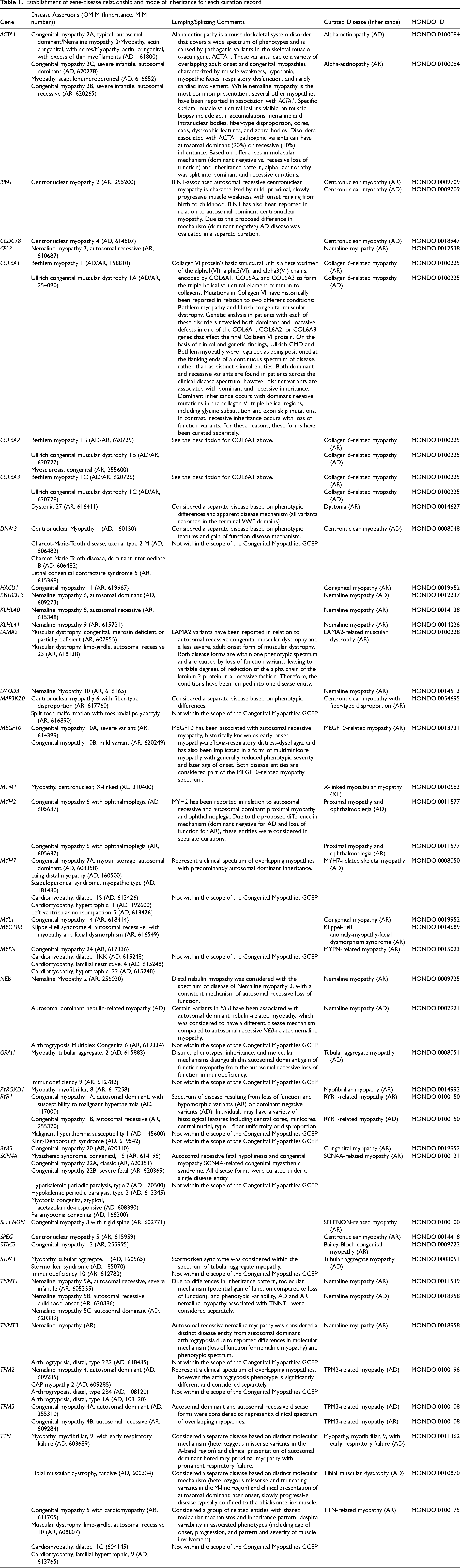
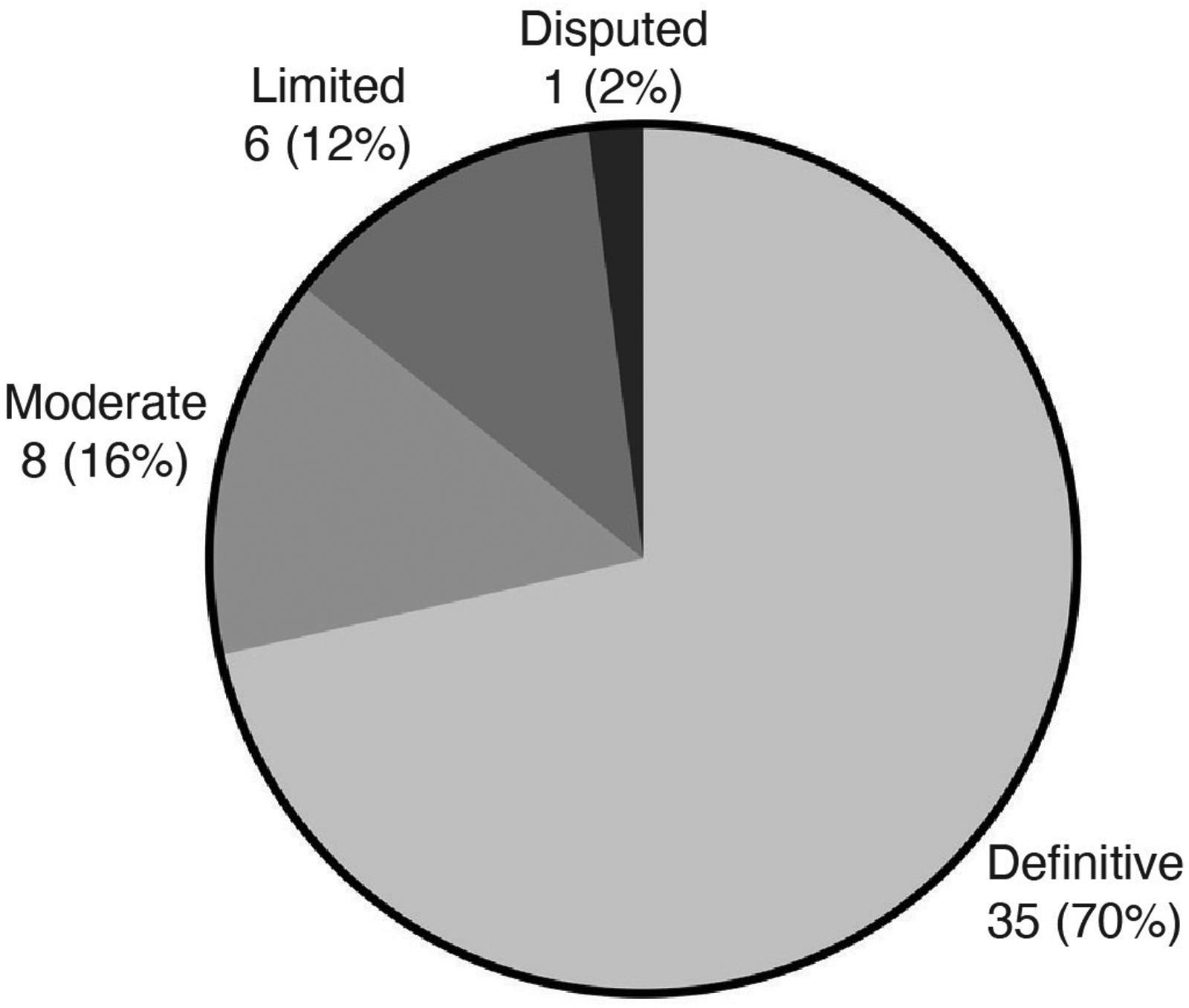
Overall, the CongenMyopathy-GCEP curated 37 genes for 50 gene-disease relationships, which resulted in 35 (70%) Definitive, eight (16%) Moderate, six (12%) Limited, and one (2%) Disputed disease relationship classifications (Figure 1, Table 2). Those classified as Definitive were scored 12–18 points. Twelve classifications, including well-established disease genes such as ACTA1, NEB, and MTM1, reached the maximum score of 18 (Figure 2). Genetic evidence reflecting numerous reports from the literature of causative variants reached the maximum of 12 points in 28 (80%) of the Definitive classifications and 56% of all classifications. All genes with 12 points of genetic evidence were classified as having Definitive evidence for the respective diseases. Additional experimental evidence was available for all Definitive classifications. The experimental evidence reached the maximum score of 6 points in 14 (40%) of the Definitive classifications and 28% of all classifications, including KLHL41, which was classified as having Moderate evidence for nemaline myopathy due to having 2.8 points for genetic evidence at the time of curation. There were no gene-disease relationships classified as Strong, due to the absence of curated genes with a total of ≥12 points without replication over time, which is the only criteria that distinguishes Strong from Definitive classification.
Clinical validity classifications for genes implicated in congenital myopathies.
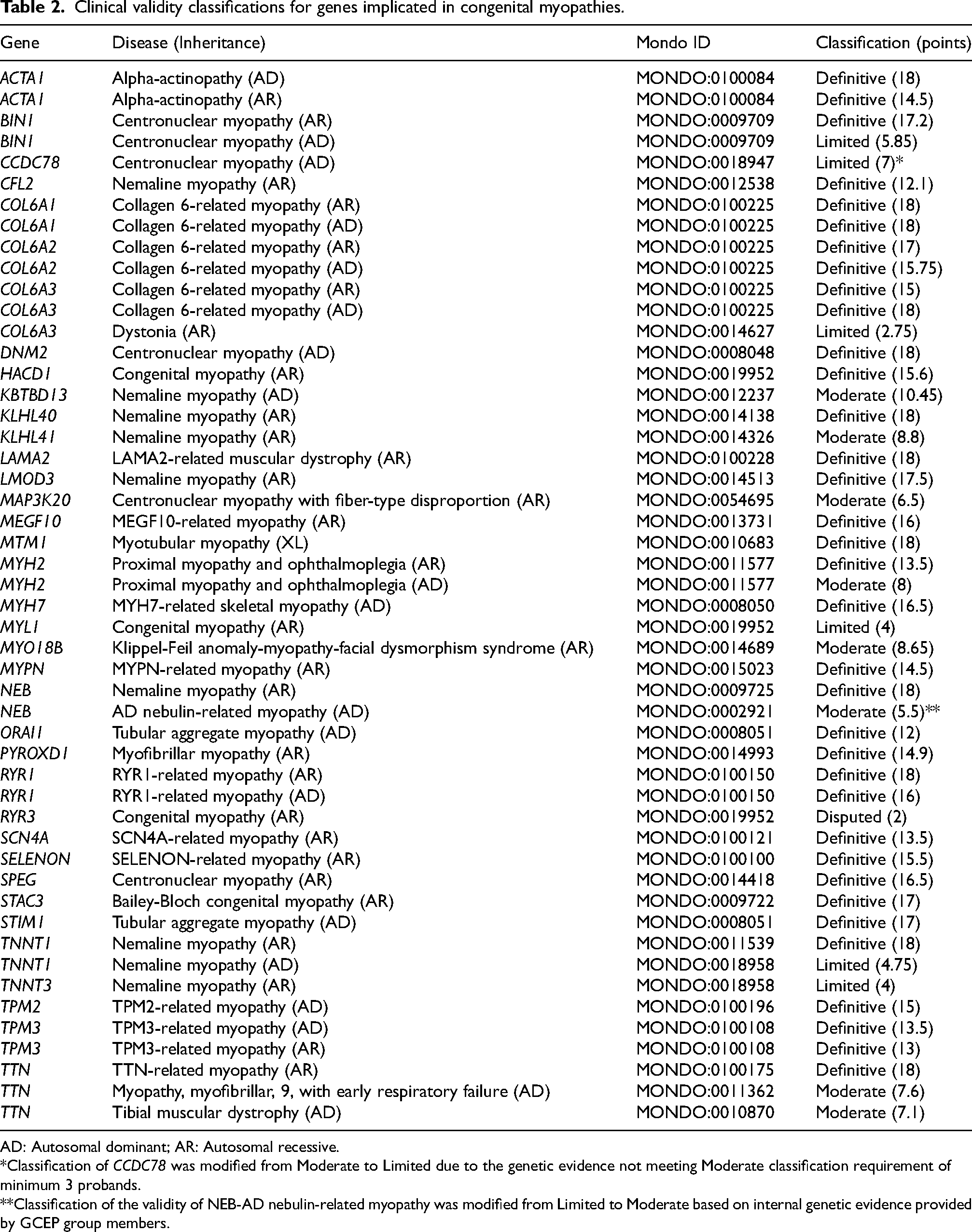
AD: Autosomal dominant; AR: Autosomal recessive. *Classification of CCDC78 was modified from Moderate to Limited due to the genetic evidence not meeting Moderate classification requirement of minimum 3 probands. **Classification of the validity of NEB-AD nebulin-related myopathy was modified from Limited to Moderate based on internal genetic evidence provided by GCEP group members.
Non-Definitive classifications earned 2–10.45 points and lacked evidence in genetic and/or experimental categories (Figure 2). Gene-disease relationships that reached a higher classification had a higher number of probands that reached a higher genetic evidence score. KBTBD13, KLHL41, MAP3K20, MYH2, MYO18B, NEB-related AD nebulin-related myopathy, TTN-related AD myofibrillar myopathy with early respiratory failure, and TTN-related AD tibial muscular dystrophy were classified as having Moderate evidence for gene-disease association. Current evidence of a relationship with congenital myopathies was considered as Limited for CCDC78 for centronuclear myopathy and MYL1 with congenital myopathy. CCDC78 had a total score of 7 points, which would have been sufficient for Moderate disease relationship classification; however, only 2 probands with CCDC78 variants were available to be scored as genetic evidence. Due to the requirement for a minimum of 3 probands for Moderate classification, the relationship of CCDC78 with centronuclear myopathy remained as Limited at this time. Four genes that have been implicated in multiple diseases were also considered to have Limited evidence for a relationship with congenital myopathies: BIN1 association with AD centronuclear myopathy, TNNT3 association with AR nemaline myopathy (relationship with AD distal arthrogryposis was not considered), TNNT1 with AD nemaline myopathy, and COL6A3 with AR dystonia. Gene-disease relationships with Limited validity had two or fewer probands reported at this time.
Finally, RYR3 was classified as having a Disputed disease relationship. RYR3 was reported once in relation to AR congenital myopathy in a single affected individual in 2018. 11 The two unique reported missense variants have been the only variants asserted to be associated with disease and the impact of these variants on protein function is currently unclear. Some functional evidence supporting this gene-disease relationship exists, including protein expression data, 16 functional alteration data from Ryr3 knockout mice,17,18 and RYR3-depleted zebrafish. 19 Although the relevant experimental evidence is supportive of a gene-disease relationship, because the single report implicating RYR3 in AR congenital myopathy in humans was considered as being insufficient to be scored as genetic evidence at this time, the relationship of RYR3 to congenital myopathies was classified as Disputed.
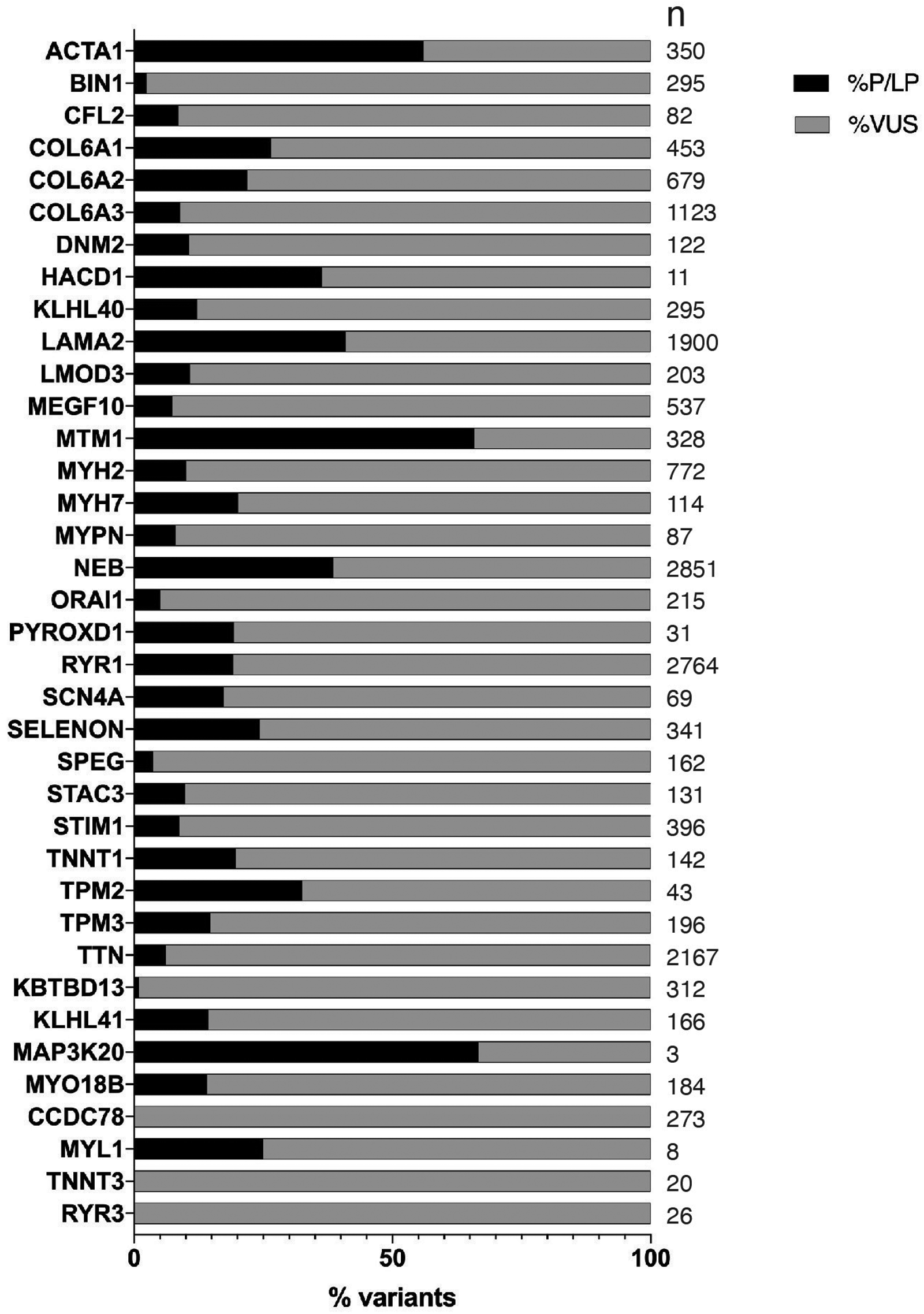
To assess how gene-disease relationship classifications may impact variant classifications in congenital myopathy genes, we analyzed the variant classifications that have been submitted to ClinVar for each gene. In ClinVar, variants in genes with non-Definitive roles with submitted classifications for congenital myopathy-related conditions were predominantly variants of uncertain significance (VUS). In genes with non-Definitive classification, only 5.7% (57/992) of variants other than those classified as benign/likely benign were classified as pathogenic or likely pathogenic (P/LP), whereas 94.3% were classified as VUS (1471/1564), with 99.3% (299/301) of such variants classified as VUS in genes with Limited classification and 91.7% (610/665) classified as VUS in genes with Moderate classification. On the other hand, in genes with a Definitive role in disease, 22.6% (3789/16769) of variants other than benign/likely benign were classified as P/LP and 77.4% were classified as VUS (Figure 3). Three genes curated by the CongenMyopathy-GCEP (CCDC78, RYR3, and TNNT3) did not have any P/LP ClinVar submissions related to congenital myopathies that met criteria for analysis, all variants in these genes were classified as VUS by the submitters.
Discussion
A substantial proportion of genes reported in patients with congenital myopathies have been associated with a wide phenotypic spectrum and overlapping features, which poses a challenge in the molecular diagnosis of patients. Although genetic testing provides the opportunity to make a molecular diagnosis for genetically heterogenous conditions, its utility is often limited by inconsistency and uncertainty in the validity of a gene's role in disease and the clinical significance of genetic variants.
Of the 50 gene-disease relationships that were curated by the CongenMyopathy-GCEP, 70% had Definitive level of evidence for a causal role in one or more types of congenital myopathies. Gene-disease validity classifications have been published by other ClinGen GCEPs investigating various genetic disorders.20–31 The proportion of curated genes classified as having Definitive evidence for disease relationship ranges from 5% (Brugada syndrome) to 75% (intellectual disability/autism).23,30 Many factors may contribute to the relative proportion of genes with non-Definitive classification in different disease groups, including the ambiguity and complexity of disease phenotypes or presence of reliable functional studies and animal models.
Curation of distinct phenotypes and inheritance patterns is important to enable a better prediction of the possible disease presentation and inheritance patterns when novel variants are identified in these genes. Eleven of the 37 genes curated were determined to be associated with more than one distinct congenital myopathy subtype due to differences in molecular mechanism, phenotype, and/or inheritance pattern (Table 1). For example, RYR1 was evaluated separately for AD and AR inheritance, based on the difference in pathogenic mechanisms (dominant negative for AD versus loss of function for AR), and phenotypic variability, most notably the overlap of the AD phenotype with malignant hyperthermia susceptibility. On the other hand, TNNT1 was evaluated separately for the classically observed association of loss of function variants with AR nemaline myopathy and the distinctive phenotypic features not being present in the more recently defined association of potential gain of function variants with AD nemaline myopathy. 27 The strength of a gene's associations with different diseases was variable. For example, the causal role of COL6A3 in collagen 6-related myopathy was classified as Definitive, while its relationship with dystonia was considered as having only Limited level of evidence. The gene curated by the CongenMyopathy-GCEP related to the highest number of disease entities was TTN. Multiple diseases, including cardiomyopathies, muscular dystrophies, and myopathies have been associated with TTN variants in the literature. In particular, the TTN-associated neuromuscular disorders encompass a range of distinct clinical presentations with wide variation in severity, age of onset, progression, and pattern of muscle involvement, as well as differences in inheritance patterns and suspected molecular mechanisms. Because of these differences, the curation of this gene was separated into three distinct gene-disease pairs to allow individual evaluation of the unique disease entities (Table 1). The size of the TTN gene and the encoded protein, as well as the number of diverse isoforms complicate functional studies on this gene. As a result, none of the TTN curations reached the maximum number of points for experimental evidence. For example, although over twelve TTN missense variants have been associated with AD myofibrillar myopathy with early respiratory failure32–37 to date, including three recurrent variants identified in multiple affected individuals, lack of sufficient experimental evidence supporting the effect of the variants on protein function led to this gene-disease relationship being classified as having Moderate evidence. Conversely, a broad spectrum of congenital myopathy phenotypes was associated with four of the genes classified as having Definitive evidence (ACTA1, COL6A1, COL6A2, and COL6A3). For these genes, disease names that encompass the continuum of histological and clinical phenotypes were selected, rather than the existing disease names that are commonly used for referring to the distinct clinical or pathological presentations. Thus, ACTA1 was curated for the broad disease entity of alpha-Actinopathy, which includes overlapping congenital and adult-onset myopathies characterized by muscle weakness, hypotonia, myopathic facies, respiratory dysfunction, and rarely, cardiac involvement. Inter- and intra-familial phenotypic variability observed for individual variants suggested that these entities are part of the same clinical spectrum. While nemaline myopathy is the most common presentation, several other histopathologically distinct myopathies have been reported in association with ACTA1 variants. Specific skeletal muscle structural lesions visible on muscle biopsy include actin accumulations, nemaline and intranuclear bodies, fiber-type disproportion, cores, caps, dystrophic features, and zebra bodies. 38 Approximately 200 unique variants have been reported to date in ACTA1, resulting in both dominant (∼90%) and recessive disease (∼10%). 38
Of note, 30% (15 of 50) of gene-disease associations have Moderate, Limited, or Disputed classification. Six gene-disease relationships were classified as having Limited evidence. All Limited classifications, except for CCDC78, are for relatively recent disease gene discoveries and although more genetic and/or experimental evidence is needed to support a causal role, no convincing evidence has emerged that contradicts the proposed gene-disease relationships.
A Moderate classification typically indicates that there is emerging but insufficient evidence for the validity of gene-disease relationship at the time of curation to classify as Strong, although it is likely to reach Strong/Definitive in time. 39 Moderate classifications will be re-evaluated every two years to maintain current gene-disease validity classifications and may become Strong and Definitive classification over time as evidence accrues. 39 Updates to gene-disease validity classifications will be published on the ClinGen CongenMyopathy-GCEP website. Whether the genes with Limited level of evidence will have further data supporting a higher disease validity classification in the future depends on the availability of additional case and experimental studies. To improve understanding of gene- and variant-disease relationships, we encourage publication of additional studies particularly for the genes with non-Definitive relationships. The importance of available case reports is exemplified by two genes, HACD1 and MYO18B, which were reclassified from having Limited to Moderate or Definitive evidence, respectively, based on recently published studies during the CongenMyopathy-GCEP's four-year working period. Two additional genes with Moderate classification, KBTBD13 and KLHL41, each have strong experimental evidence (5.5–6 points) and could be reclassified to having higher validity if additional cases are reported in the future. Data have shown that many genes in the Limited category, particularly those that remain Limited for more than five years, do not accumulate evidence in the future to move to a higher classification. 39 However, the assembly of additional supportive case series for such rare genetic conditions typically takes considerable time, so that additional publications for these genes may still emerge in the future, allowing for the classification to be adjusted to a higher level of evidence.
Interpretation of a variant's clinical significance is dependent on the understanding of the gene's relationship with disease. This study presents the evaluation of the first 37 genes that have been curated by our group. Our gene curation efforts continue addressing new genes that are implicated in congenital myopathies and will be ongoing as new gene-disease relationships are published in the future. We will continue to share our curations and classifications on the publicly available ClinGen website as they get completed. In our analysis, genes with a non-Definitive role in congenital myopathies had a higher proportion of variants classified as VUS compared to genes with Definitive roles. Evidence-based curation of genes is essential to guide appropriate classification and reporting of variants and is expected to improve variant interpretations to assist in patient care. Through systematic assessment of gene-disease relationships in combination with evidence supporting individual variant classifications, the CongenMyopathy-GCEP aims to facilitate accurate molecular diagnoses of patients with congenital myopathies.
Supplemental Material
sj-docx-1-jnd-10.1177_22143602251339369 - Supplemental material for Clinical validity of congenital myopathy genes determined by the ClinGen Congenital Myopathies Expert Panel
Supplemental material, sj-docx-1-jnd-10.1177_22143602251339369 for Clinical validity of congenital myopathy genes determined by the ClinGen Congenital Myopathies Expert Panel by Justyne E Ross, May Flowers, Shannon McNulty, Mayher Patel, Hui Yang, Brooke Palus, Marwa Abdelmoneim Elnagheeb, Lucy Eng, Emma Owens, Alan H Beggs, Enrico Bertini, Adele D'Amico, Sandra Donkervoort, James Dowling, Fabiana Fattori, Ana Ferreiro, Casie A Genetti, Hernan Gonorazky, Monkol Lek, Amanda Lindy, Livija Medne, Francesco Muntoni, Sander Pajusalu, Katarina Pelin, John Rendu, Anna Sarkozy, Matteo Vatta, Tom Winder, Grace Yoon, Carsten G Bönnemann and Ozge Ceyhan-Birsoy in Journal of Neuromuscular Diseases
Supplemental Material
sj-docx-2-jnd-10.1177_22143602251339369 - Supplemental material for Clinical validity of congenital myopathy genes determined by the ClinGen Congenital Myopathies Expert Panel
Supplemental material, sj-docx-2-jnd-10.1177_22143602251339369 for Clinical validity of congenital myopathy genes determined by the ClinGen Congenital Myopathies Expert Panel by Justyne E Ross, May Flowers, Shannon McNulty, Mayher Patel, Hui Yang, Brooke Palus, Marwa Abdelmoneim Elnagheeb, Lucy Eng, Emma Owens, Alan H Beggs, Enrico Bertini, Adele D'Amico, Sandra Donkervoort, James Dowling, Fabiana Fattori, Ana Ferreiro, Casie A Genetti, Hernan Gonorazky, Monkol Lek, Amanda Lindy, Livija Medne, Francesco Muntoni, Sander Pajusalu, Katarina Pelin, John Rendu, Anna Sarkozy, Matteo Vatta, Tom Winder, Grace Yoon, Carsten G Bönnemann and Ozge Ceyhan-Birsoy in Journal of Neuromuscular Diseases
Footnotes
ORCID iDs
Funding
This publication was supported by the National Human Genome Research Institute of the National Institutes of Health through the following grants: U24NS120858, U41HG009650, U24HG009650, and U24HG006834. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Declaration of conflicting interests
M.V. receives salary from Labcorp Genetics Inc. (formerly Invitae Corp.); E.B. received reimbursements for Advisory Board by Biogen, PTC, Novartis, Roche, and Pfizer; A.H.B. has received compensation or honoraria from Audentes Therapeutics, F. Hoffman-LaRoche AG, GLG Inc, Guidepoint Global LLC, and Kate Therapeutics Inc; and holds equity in Kinea Bio and Kate Therapeutics Inc; A.S.L. is employed by, and maintains stock ownership or options with, GeneDx, LLC.
Data availability
All curation summaries and relevant references are available at https://search.clinicalgenome.org/kb/affiliate/10031.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.