
Abstract
Background:
Whole-exome sequencing (WES) facilitates the diagnosis of hereditary neuromuscular disorders. To achieve an accurate diagnosis, physicians should interpret the genetic report carefully along with clinical information and examinations. We described our experience with (1) clinical validation in patients with variants found using WES and (2) a diagnostic approach for those with negative findings from WES.
Methods:
WES was performed on patients with the clinical impression of hereditary neuromuscular disorders. Information on clinical manifestations, neurological examination, electrodiagnostic studies, histopathology of muscle and nerve, and laboratory tests were collected.
Results:
Forty-one patients (Male/Female: 18/23, age of onset: 34.5±15.9) accepted WES and were categorized into four scenarios: (1) patients with a positive WES result, (2) patients with an inconclusive WES result but supporting clinical data, (3) negative findings from WES, but a final diagnosis after further work-up, and (4) undetermined etiology from WES and in further work-ups. The yield rate of the initial WES was 63.4% (26/41). Among these, seventeen patients had positive WES result, while the other nine patients had inconclusive WES result but supporting clinical data. Notably, in the fifteen patients with negative findings from WES, four patients (26.7%) achieved a diagnosis after further workup: tumor-induced osteomalacia, metabolic myopathy with pathogenic variants in mitochondrial DNA, microsatellite expansion disease, and vasculitis-related neuropathy. The etiologies remained undetermined in eleven patients (myopathy: 7, neuropathy: 4) after WES and further workup.
Conclusions:
It is essential to design genotype-guided molecular studies to correlate the identified variants with their clinical features. For patients who had negative findings from WES, acquired diseases, mitochondrial DNA disorders and microsatellite expansion diseases should be considered.
INTRODUCTION
Neuromuscular disorders (NMDs) are a clinically and genetically heterogeneous group of disorders affecting peripheral nerves or muscles. Over the past decades, next-generation sequencing (NGS) has revolutionized the diagnostic paradigm in genetic neuromuscular disorders, and whole-exome sequencing (WES) is one of these powerful diagnostic tools. WES investigates protein-coding genes, approximately 3% of the whole genome, and the protein-coding genes accounts for up to 60% of disease-causing genomic variation [1]. The comprehensive interpretation of clinical manifestations, pathology, electrophysiological studies, and WES reports leads to accurate diagnosis of NMDs.
Previous studies have shown that the early utilization of WES facilitates more rapid and accurate NMD diagnosis, especially in monogenetic diseases [2–4]. The overall diagnosis yield rate ranges from 40–60% in myopathic patients and 30–40% in neuropathy patients, with a higher yield rate in pediatric patients and a lower yield rate in single proband studies [5–10]. The negative-exome results may be caused by somatic mosaicism, copy number variants, mitochondrial mutations, structural variants not properly captured through exome sequencing, and secondary causes not identified [11].
For exome-positive cases with known pathogenic variants that had been well investigated, the final diagnosis can be reached using phenotype-genotype correlations. However, careful interpretation along with further evaluation are essential for exome-positive cases without previously reported pathogenic variants and exome-negative cases [12]. Herein, we described our experience in integrating WES into a diagnostic approach in patients with NMDs. We focus on (1) advanced molecular tests in patients with variants found using WES and (2) a diagnostic approach for those with negative findings from WES.
METHODS
Patient recruitment
This retrospective cohort study (202109079RIBN) was approved by the Ethics Committee of the National Taiwan University Hospital (NTUH), Taipei, Taiwan. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. The inclusion criteria were: (1) an impression of hereditary NMDs, (2) no other etiology identified after selective genetic testing before the WES study, and (3) clinical data were available. We arranged WES to all the probands, and for the proband’s family, Sanger sequencing on the specific variants revealed by the proband-only WES was arranged if they agreed.
Clinical evaluations, laboratory study, and electrophysiological examinations
The clinical data, including age of disease onset, sex, and pedigree, were collected in all patients. In patients with myopathy, we additionally collected the serum creatinine kinase (CK) levels and the informations of muscle histopathology. According to previous studies, distribution of weakness and associated symptoms can be categorized into different patterns for clinical diagnosis. The presentation of our patients was categorized as follows: (1) limb-girdle weakness, (2) scapuloperoneal weakness, (3) distal arm and proximal leg weakness, (4) distal weakness, (5) ptosis with or without ophthalmoplegia, (6) episodic weakness/myalgia/rhabdomyolysis, and (7) asymptomatic hyperCKemia. The muscle histopathology was evaluated by the neurologist and pathologist together. Routine histochemical staining in our hospital encompassed hematoxylin and eosin staining, Gomori-Trichome staining, succinate dehydrogenase staining, nicotinamide adenine dinucleotide staining, and staining for myosin adenosine triphosphatase at pH 4.3 and 10.42. Based on clinical information and routine histochemistry results, specific immunohistochemistry would be arranged. Electron microscopy studies of muscle biopsy were also performed on some samples.
In patients with polyneuropathy, we collected clinical data and performed nerve conduction study (NCS). We used the electrophysiological criteria for chronic inflammatory demyelinating polyradiculoneuropathy to define demyelination and axonal loss.
Whole-exome sequencing
WES was performed using an exome capture kit probe (Agilent V6 (before Jan 2021) and Roche Hyperexome (after Feb 2021)). We then did sequencing with a NovaSeq6000 sequencer (Illumina). The protein-coding genes and flanking intronic sequences (50 bp) were studied with a 300-bp paired-end run and an average of > 150-fold coverage.
The human reference genome is GRCH38 and we used the Burrows–Wheeler Aligner (BWA) for sequence alignment and used the Genome Analysis Tool Kit (GATK V3.5, Broad Institute) [13] for variant calling. Variants were first annotated using ANNOVAR (http://wannovar.wglab.org/) [14]. Then, the annotated profiles were tagged with information of the inheritance pattern from OMIM (https://omim.org/), variant pathogenicity from ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and the allele frequency from the Taiwan Biobank (https://taiwanview.twbiobank.org.tw/). The variant filtering criteria incorporated maximal minor allele frequency (included if < 0.01) and in-silico analysis of variant severity (nonsynonymous variant, nonsense, deletion, splicing, insertion, or splicing). We manually calssified the pathogenicity of variants according to American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology guidelines [15–19]. We further defined the results of WES as “positive”, “inconclusive” and “negative” based on ACMG criteria. The positive results were defined as the variants that had been asserted as pathogenic or likely pathogenic in ACMG criteria and definite genetic diagnosis according to the inheritance pattern of gene. That is, for those compound heterozygous variants, only both alleles showed pathogenic or likely pathogenic in ACMG criteria were defined as “positive”. The inconclusive results were those being classified as variants of uncertain significance in ACMG criteria. The results of the WES were defined as negative if (1) no possible culprit variant identified in the WES or (2) the identified variants were excluded by the segregation analysis. For copy number analysis (CNV), with SAM/BAM file as input, the GATK germline CNV (gCNV) caller can generate a VCF file containing the CNVs found during the variant calling process [20].
Clinical validation of WES results: Clinical-Molecular diagnosis
In patients with myopathy, the pathogenicity was defined as “definite” in two situations: (1) patients presented with the representative clinical phenotype with positive WES results, or (2) those who had a typical phenotype but with inconclusive WES results, the diagnosis validated by typical pathognomonic findings in clinical manifestations, pathology, or other examinations. Notably, there should be negative results for the other known genes with the similar phenotype in both situations [21]. Instead, pathogenicity was defined as “probable” if the criteria were not met. Among the patients with neuropathy or motor neuron disease, the clinical-molecular diagnosis was regarded “definite” only if the clinical manifestations, culprit variants, and electrophysiological study were consistent with the literature. Instead, pathogenicity was defined as “probable” if the above criteria were not met. For those with two allelic variants, the definite diagnosis required that compound heterozygous variants were confirmed. The final clinical-molecular diagnosis was made based on the genotype-phenotype correlation and was referred to Online Mendelian Inheritance in Man (OMIM) database.
RESULTS
In total, we recruited 41 patients with neuromuscular disorders who accepted the WES study. Most of the patients (n = 28) had a clinical diagnosis of myopathy, while the other 10 patients were clinically diagnosed with neuropathy, and 3 patients were diagnosed with motor neuronopathy. The clinical phenotypes, laboratory findings including electrophysiological and pathology, and genetic findings are summarized in Tables 1, & 4, Fig. 1, and supplementary. Overall, twenty-six patients (63.4%) had a likely diagnosis from the WES, meeting the clinical-molecular diagnosis [21], most of whom were diagnosed with myopathy, except four patients who were diagnosed with neuropathy or neuronopathy. Eight cases had biallelic variants, and compound heterozygosity was confirmed in four cases. Five patients could not meet the “definite” clinical-molecular diagnosis due to lack of segregation analysis. There were four scenarios among these 41 patients:
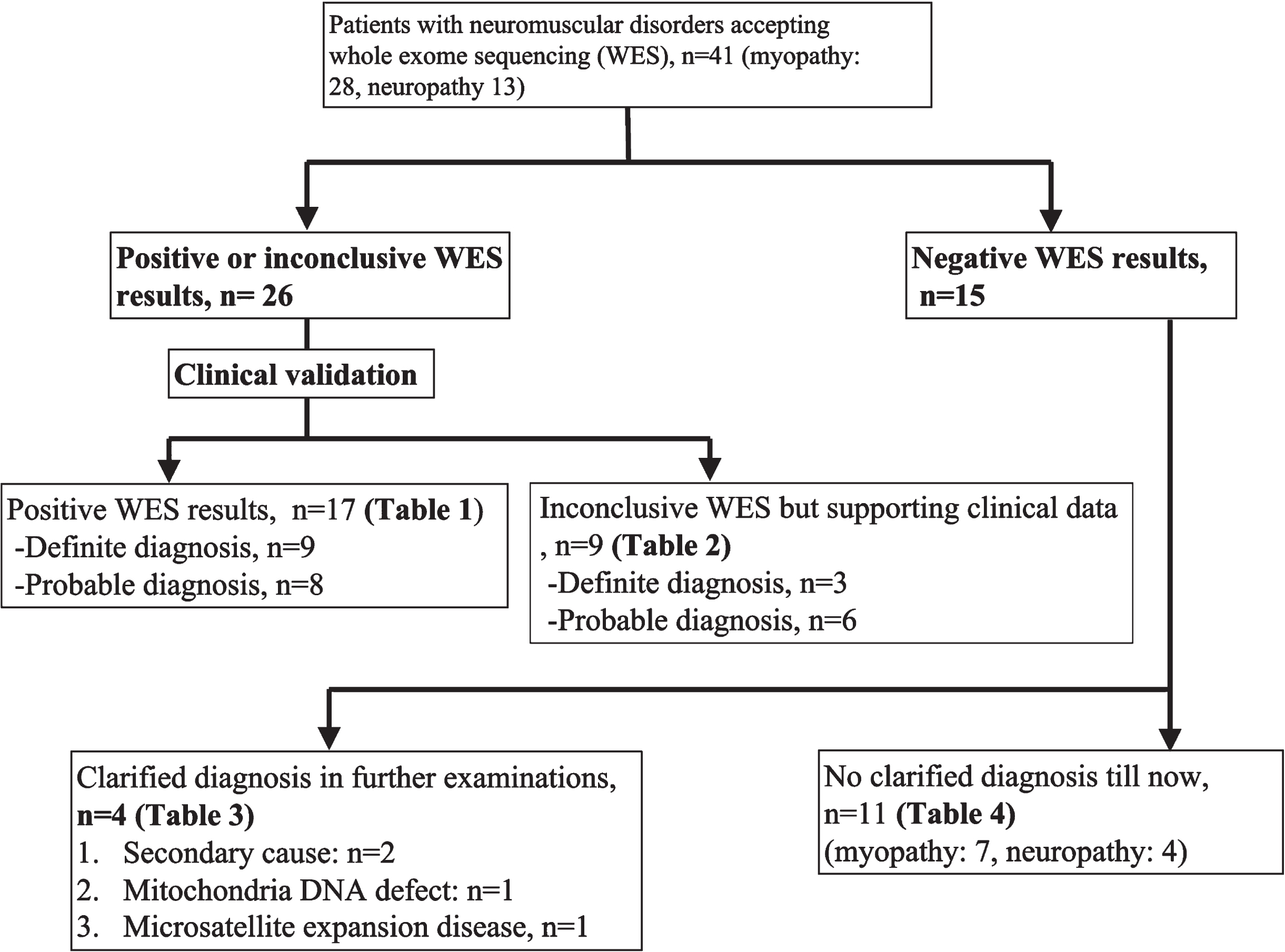
Algorithm of the study.
Patients with a positive whole-exome sequencing result
Patients with an inconclusive whole-exome sequencing result but supporting clinical data
Patients with negative WES result, but with final clinical diagnosis after further work-up
Scenario 1: Patients with a positive WES result
Among these 17 patients with positive WES result (Table 1), most presented with a typical clinical phenotype. We made the diagnosis with genotype-phenotype correlation. In the group of definite diagnosis, two cases with atypical presentations (Cases 6 and 7). Case 6 was a male with exercise intolerance and high hyperCKemia. The work-up of metabolic myopathy and mitochondrial myopathy was normal. However, the patient had partial loss of dystrophin staining in muscle biopsy, supporting the diagnosis of dystrophinopathy. Case 7 had hand weakness as an initial symptom but later developed lower limb distal weakness, which is rarely reported in GNE myopathy.
There were 8 patients classified as members of the probable diagnosis group (myopathy: 5, neuropathy: 3). Three patients (CAPN3, TTN, and SCN4A) with atypical or uncommon clinical phenotypes could not meet the definite diagnosis criteria, and segregation analysis and molecular studies were needed. Four patients (FHL1, FBXO38, POLG, and OPA1) had variants that had been asserted as pathogenic or likely pathogenic in ClinVar database, and one patient (DYSF) need a segregation analysis to ascertain compound heterozygote.
Scenario 2: Patients with an inconclusive WES result but supporting clinical data
There were 9 patients showing inconclusive WES result but having supporting clinical data. Of note, 3 patients with myopathy who carried the new variants fulfilled the proposed definite diagnosis criteria. Their diagnosis was validated by (1) typical pathognomonic findings yielded from the clinical manifestations or muscle biopsy (e.g., distal myopathy and atrophic myocytes with desmin aggregates in DES-related myofibrillar myopathy 1; limb-girdle weakness with partial loss of dystrophin staining in muscle biopsy), or (2) further molecular examinations (e.g., mitochondria DNA sequencing and muscle electron transfer chain activity assay in TK2-related myopathy [22]).
There were another 6 patients classified in the probable diagnosis group (myopathy: 3, neuropathy: 3). Three patients (SLCO2A1, POLG, and SCO2) with representative clinical phenotypes could not meet the definite diagnosis criteria due to the lack of segregation analysis to ascertain compound heterozygote. Notably, in a patient with SLCO2A1-related hypertrophic osteoarthropathy we found a nonspecific myopathic change in the proximal muscles, which has not been previously reported. The patient with variant in TTN gene had early respiratory failure when she could still ambulate. However, we classified the case as a probable diagnosis because the variant was not located in the common region related to hereditary myopathy with early respiratory failure (HMERF). Although the patient had no necklace body in the muscle pathology, the abnormal desmin and dystrophin subsacrolemmal in light microscopy and Z-disk alterations in electron microscopy are compatible with the pathological diagnosis of myofibrillar myopathy, which has been reported in TTN-related HMERF [23, 24]. Two patients had variants in the NEFH gene. We made the diagnosis of brachial amyotrophic diplegia, primary lateral sclerosis, neuronopathy, distal hereditary motor neuropathy, and type IID based on the clinical presentation, electrophysiological studies, and WES results.
Scenario 3: Patients with negative WES result, but with final clinical diagnosis after further work-up
Notably, in the 15 patients without a molecular diagnosis from WES, four patients (26.7 %) had a diagnosis after further work-up. The details of these four patients are described below.
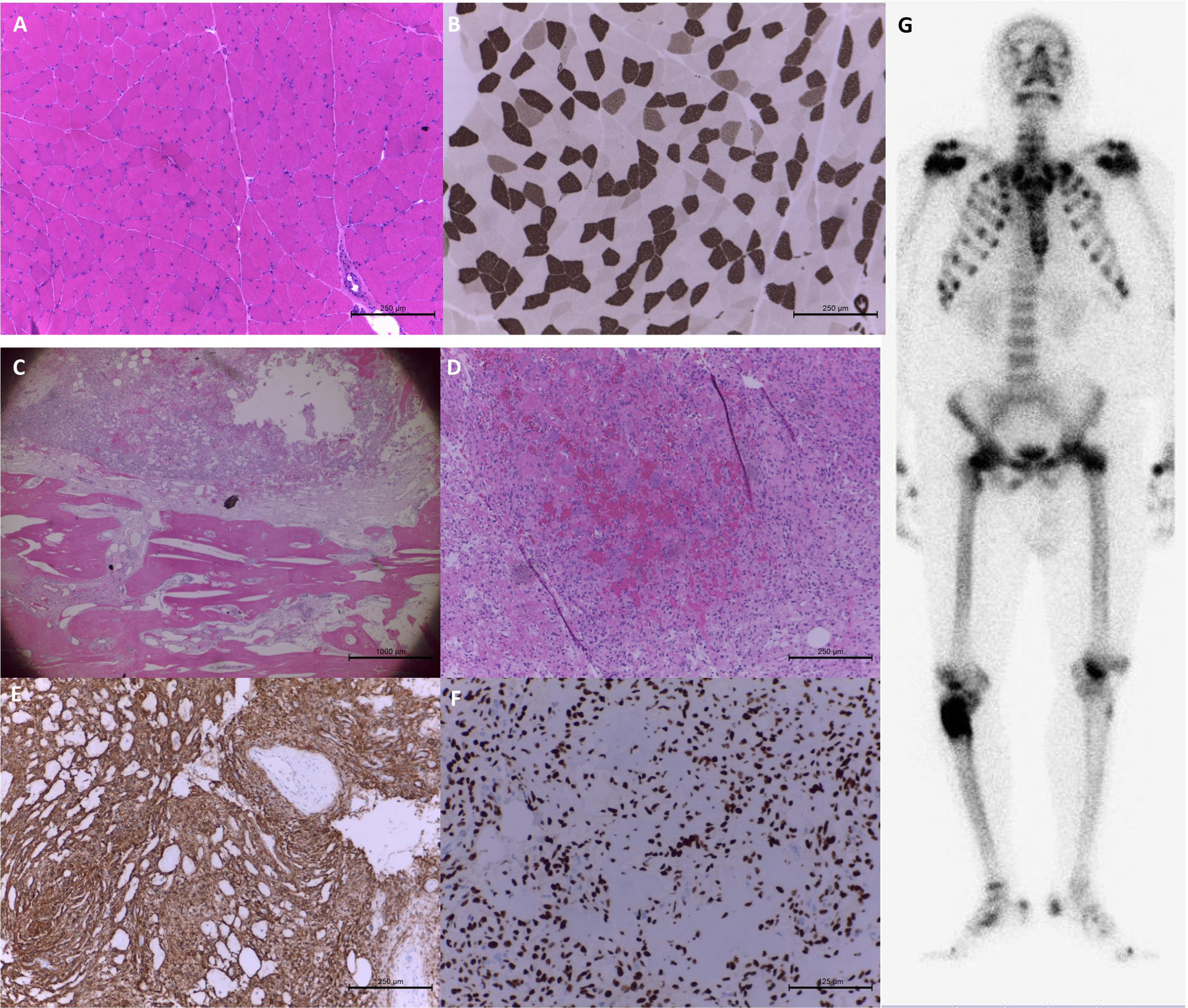
Muscle and bone histopathology in patient 28 with tumor-induced osteomalacia. The staining of hematoxylin and eosin (H&E) (A) and myosin adenosine triphosphatase at pH 4.3 (B) showed compacted muscle fibers with scattered atrophic fibers in the biopsy at left biceps brachii. The bone tumor pathology showed spindle tumor cells in myxohyalinized fibrotic stroma with eosinophilic matrix-like materials accompanying the bony components in H&E staining. (C&D) Focal bony destruction, woven bone formation and grungy calcification were also noted. The immunohistochemical staining of CD56 (E) and SATB2 (F) was strongly positive. Whole-body bone scintigraphy revealed a bone tumor with markedly increased osteoblastic activity at the right proximal femur. (G) Additionally, there were multiple posttraumatic changes in the thoracic cage, pubic bones, and bilateral femurs.
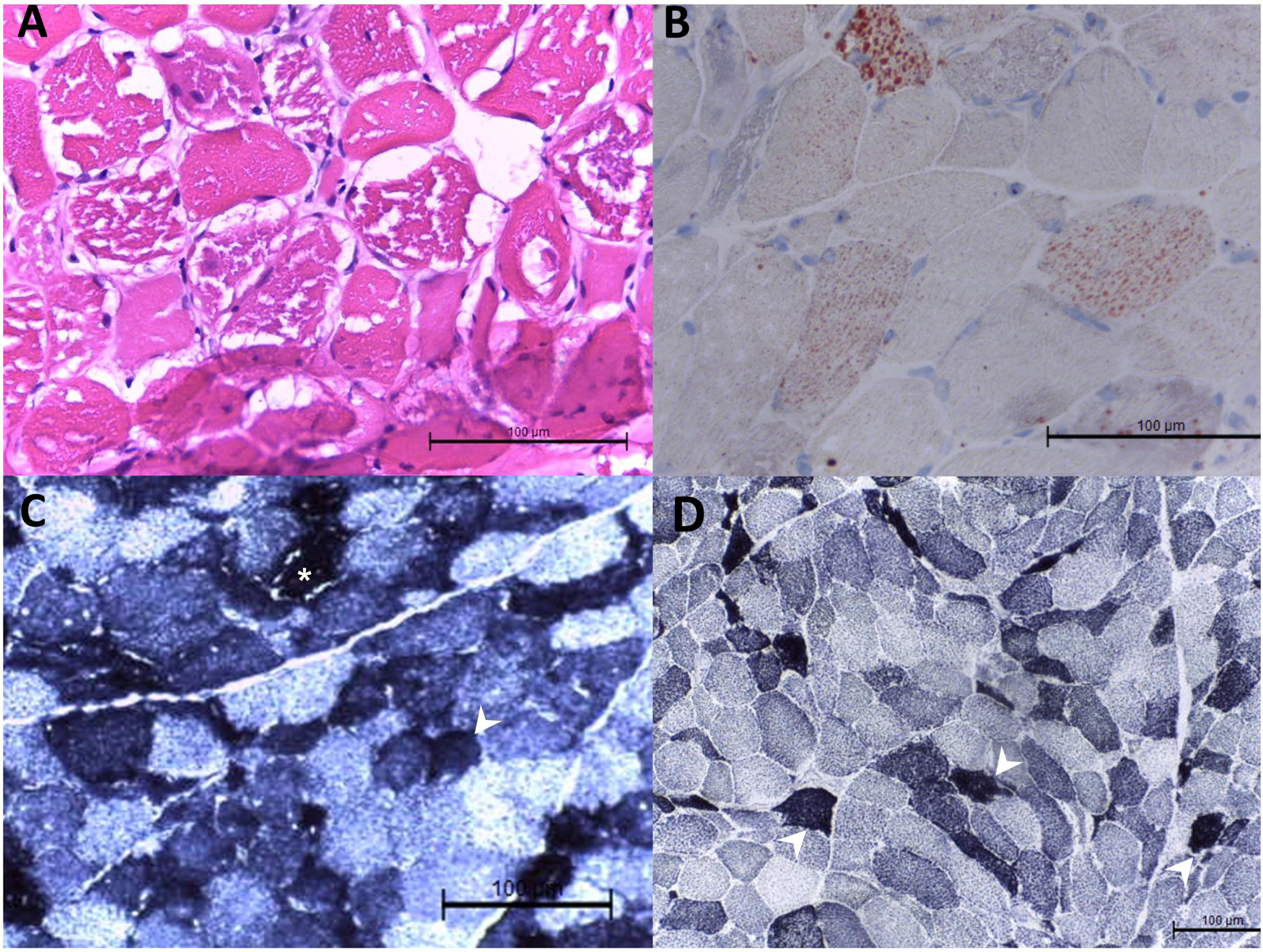
Muscle histopathology in patient 29 with mitochondrial myopathy. Hematoxylin and eosin staining showed variated muscle fiber sizes, degenerative and atrophic muscle fiber sizes, and nuclear internalization in the biopsy at the left biceps brachii. There were muscle fibers with variated cytoplasmic vacuoles. The vacuole was strongly positive for Oil Red O staining (B). In the staining of nicotinamide adenine dinucleotide-tetrazolium reductase (NADH-TR) (C) and succinate dehydrogenase (D), dark staining was noted in some muscle fibers (arrowhead). Vacuoles were also seen in the NADH-TR staining ( *).
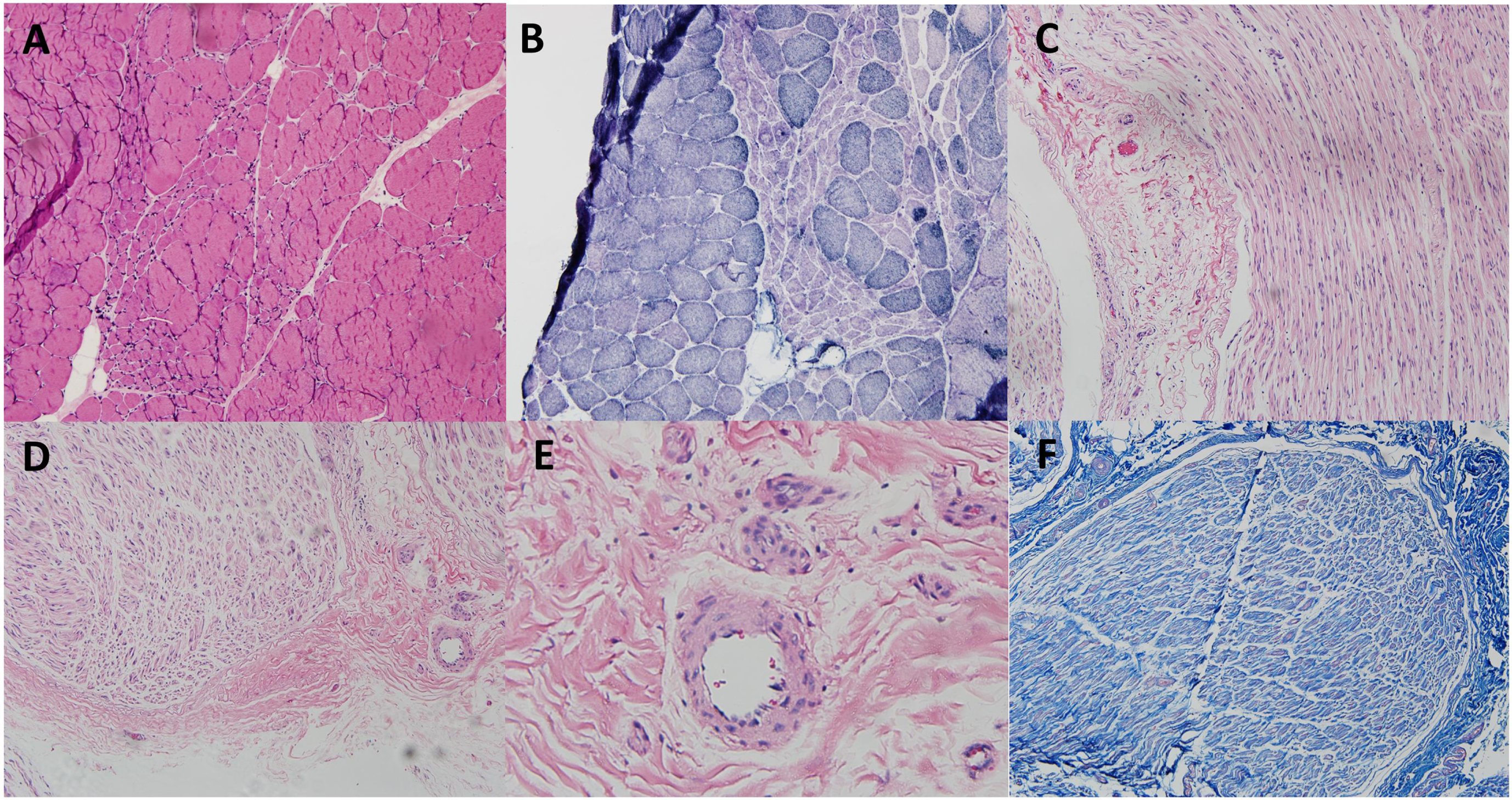
Combined muscle and nerve biopsy in patient 31. The staining of hematoxylin and eosin (H&E) (A) and nicotinamide adenine dinucleotide-tetrazolium reductase (B) showed grouped atrophy in the right peroneal brevis muscle. The biopsy in the right superficial peroneal nerve did not show thickening of small vessels, lymphocyte infiltration, red blood cell extravasation in H&E (C-E) and Masson’s trichrome staining (F).
Scenario 4: Patients with negative WES result and undetermined etiology in further work-ups
For the remaining 11 patients with a negative molecular diagnosis from WES, most (n = 7) had a clinical diagnosis of myopathy, while others (n = 4) were clinically diagnosed with neuropathy. The detailed clinical features of patients without definite diagnosis are presented in Table 4. Among these, some had negative findings from NGS. Two had suspected variants at first but were later excluded by segregation analysis. WGS may be helpful for these patients. For patients with unreported pathological variants of ANO5 and FKRP mutations, the definite diagnosis may be verified after segregation analysis and muscle biopsy with molecular studies. Currently, the patient is still hesitant about this plan. Some had suspected variants but without reported phenotypes, such as POLG-related Charcot-Marie-Tooth disease type 1 (CMT1) and ACAD9-related mitochondrial myopathy. Segregation analysis and further molecular diagnosis are needed.
Patients with negative WES result and undetermined etiology in further work-ups
DISCUSSION
WES is a powerful diagnostic approach for identifying pathogenic variants in patients with genetic diseases. Even when obtaining a positive finding from WES, the physician still needs to correlate the identified variants with the clinical phenotypes. Clinical challenges remain after obtaining a negative or undetermined WES. We focus our discussion on (1) designing molecular tests in patients with positive or inconclusive WES results and (2) a diagnostic approach for those with negative WES results.
In this study, our yield rate of WES is 63.4%, which is higher than the previous reports (yield rate of myopathic patients: 40–60% ; yield rate of neuropathy patients: 30–40% [5–10]). Our WES pipeline is same as most of the study, and reports having different pipelines such as using higher minor allele frequency < 0.02 or 0.05 as variant filtering criteria did not get higher yield rates. Several possibilities may explain this slightly higher yield rate. First, we considered the WES as the second-tier genetic test. After excluding common secondary cause, we would perform first-tier single gene test according to the deep phenotyping (e.g., D4Z4 repeats in the chromosome 4q in scapuloperoneal weakness and Ala117Ser in TTR gene in late-onset pan-modality neuropathy in Taiwanese). WES was served as the second-tier genetic test when the results of the first-tier test was negative. Second, we applied the segregation analysis with Sanger sequencing on the specific variants revealed by the proband-only WES, the cost of which is much lower than the cost of trio-WES. The procedure is mandatory for the cases with two variants but uncertain allele or the cases with only one previously unidentified variant. In our cohort, this procedure accompanied with muscle biopsy or further molecular examinations upgraded the patients with inconclusive WES result to definite clinical-molecular diagnosis in one case (Case 20), and exclude the variants as the pathogenic variants in two cases (Case 39 and 40). Of note, four patients could only meet the criteria of probable clinical-molecular diagnosis due to uncertainty of compound heterozygosity. Third, we arranged the specific genotype-guided molecular studies to investigate and confirm the proposed pathomechanism of different genetic diagnosis. The process of extensive clinical, molecular, and pathological correlation may give us additional yield [12].
For those cases with positive WES result, the final diagnosis could be reached by phenotype-genotype correlations. Interestingly, PMP22 deletion was found using WES in one patient who had negative results from extensive workups before receiving WES. In his previous genetic test, PMP22 deletion screening was performed using PCR restriction fragment length polymorphism. However, since the patient had a DNA deletion that included the restriction site, the results of gel electrophoresis may miss the deletion [25]. In Case 10, he presented weakness and hyperCKemia, and had positive family history of early respiratory failure. The TTN gene variant c.95136T>G (p.Cys31712Trp) was identified from our patient, and this variant had been reported in one Korean family with clinical features of HMERF [26]. However, our patient has not developed with respiratory failure currently and refused the muscle biopsy. He was categorized into a probable diagnosis. In summary, a validation with clinical manifestations, pathology, or other examinations is still essential in patients with positive WES results.
Among the 9 patients with inconclusive WES result, three patients (DES, DMD and TK2) achieved a final diagnosis via further pathology or molecular studies. All had a typical clinical phenotype. In case 18, the deletion from 1189 to 1206 base pairs of the DES gene is a formerly unreported new variant. With atrophic myocytes with desmin aggregates in scattered fibers in the muscle pathology, the patient was diagnosed with myofibrillar myopathy-1 according to the concordance of genotype, phenotype, and pathology [27, 28]. In addition to the typical pathognomonic findings, in case 20 with TK2 deficiency, we made a definite diagnosis using molecular examinations such as mitochondrial DNA sequencing and muscle electron transfer chain activity assays [29–31]. With a definite diagnosis, we could offer the patients more accurate genetic consultation, prognosis prediction, and the possibility of participation of clinical trial.
Not all patients or their families agreed to accept muscle biopsies or blood tests. Definite clinical-molecular diagnoses in our cohort were not achieved because of uncertain compound heterozygote (Cases 11, 21, 23, and 26), because of atypical manifestations according to literatures (Case 9, 10, and 12), because of unreported variants in the ClinVar database (Case 13 and 22) or because of current data limitations (Cases15, 16, 17, 24 and 25). For example, in Case 9, we only identified a variant in CAPN3 gene, which had been reported in patients with both autosomal dominance inheritance and autosomal recessive inheritance [32, 33]. However, the details of the phenotype of the variant with autosomal dominance inheritance was not clear in the literature [32]. Whether the single variant could lead to his symptoms, milder than typical LGMD R2, requires segregation analysis and further molecular studies. Another example is two variants of the NEFH gene noted in two patients (Cases 24 and 25) with a clinical diagnosis of ALS or probable PLS. The NEFH gene encodes the heavy neurofilament protein playing a role in intracellular transport to axons and dendrites. In molecular and animal studies, mutation of the NEFH gene affects the efficiency of survival of motor and sensory axons, while in human studies, deletions of the NEFH tail have been found in approximately 1% of sporadic motor neuron diseases [34, 35]. The above findings support the hypothesis that the NEFH variant is associated with susceptibility to ALS. Apart from the above reasons, Case 16, with a diagnosis of POLG-related parkinsonism with sensory neuronopathy, was grouped into the probable diagnosis category. The function of the enzyme encoded by POLG is the repair and replication of mitochondrial DNA. Pathogenic variants in the POLG gene may cause variable clinical manifestations including neuropathy, parkinsonism, cerebellar ataxia, epilepsy, and chronic progressive external ophthalmoplegia [36]. Although the case matched none of the POLG-related syndromes, he indeed presented with two of the main features: sensory neuronopathy and parkinsonism [37]. Clinical or tissue evidence of mitochondrial dysfunction warrants further mitochondrial DNA testing. In summary, a genotype-guided molecular studies were warranted to ascertain the pathogenicity of the identified variants.
In cases with negative WES results, further pursuit of a diagnosis is challenging. For example, in Case 27 myopathy was suspected but WES did not yield a diagnosis. Clinical observation of decreased body height and kyphosis led to a finding of osteoporosis and renal wasting hypophosphatemia. Finally, we linked right tibial tumor, renal wasting-related hypophosphatemia, muscle weakness, and osteoporosis together and arrived at an impression of tumor-induced osteomalacia. Further evaluation of serum FGF23 and tumor biopsy confirmed our diagnosis. Careful evaluation of clinical findings and searching for acquired causes should be performed before performing WES. If the WES result is undetermined, the acquired cause should be sought again.
Other than secondary causes, mitochondrial disease and microsatellite expansion disease should be kept in mind when WES is negative. Pathogenic variants in either the mitochondrial DNA or the nuclear DNA for mitochondria could lead to mitochondrial disease, but only nuclear DNA variants can be detected using WES. Therefore, it is crucial to consider mitochondrial disease in patients with maternally inherited patterns, exercise intolerance, ophthalmoplegia, or multisystemic disorders [38]. Of note, if a blood mtDNA yielded negative results, we would further arrange a muscle mtDNA test since heteroplasmy may be present. WES is incapable of detecting large-fragment copy number variations. Therefore, microsatellite expansion disease should still be put into the differential diagnosis when WES obtains negative findings. Additionally, a thorough evaluation of nerve system is essential to capture the deficits other than the peripheral nerve system. Of note, for those who had typical clinical presentation but an inconclusive pathology study, observation of treatment response could be helpful, for instance in our Case 30 with mononeuropathy multiplex.
The etiologies remained undetermined in eleven patients after WES and further workup. There are several possibilities. First, there should be secondary causes or culprit variants beyond our current knowledge that have not been evaluated. Second, under financial limitations, we checked mitochondrial genes only when clinically highly suspect. Although we did our best to evaluate all potential patients who had mitochondrial disease via detailed history taking, lactate exercise tests, muscle biopsies, and so on, a likelihood remains of missing patients with atypical phenotypes. Finally, the technological limitations of WES prevented us from detecting pathogenic variants in introns. Whole-genome sequencing or transcriptome studies in affected tissues may help to reach a final diagnosis for these patients.
Our study had limitations. First, this is a single-center study with a small cohort. Second, we did not set up a standard protocol for WES arrangement. To reduce selection bias, we recruited patients from only two attending physicians who reached a consensus about the inclusion criteria. Third, our study did not include pediatric patients. Expanding our findings to pediatric patients would require further study. Finally, the diversity of molecular studies for different genes was unavailable and resulted in many patients being placed into the probable diagnosis category.
CONCLUSION
WES facilitates the diagnosis of hereditary neuromuscular disorders. However, it is essential to pursue further molecular studies to correlate identified variants with clinical features. For patients who had negative findings from WES, secondary causes such as mitochondria and microsatellite expansion disease should be considered. Moreover, observation of treatment response, segregation analysis and further molecular evaluation should be beneficial to the final diagnosis.
DECLARATIONS
Consent publication
All authors have read and agreed the contents of this manuscript submitted to Journal of Neuromuscular Diseases.
AVAILABILITY OF DATA AND MATERIAL
The datasets generated during analysis and/or during the current study are available from the corresponding author on reasonable request.
COMPETING INTERESTS
None.
FUNDING
None.
AUTHOR’S CONTRIBUTIONS
All authors contributed to data acquisition and analysis. P.-S.C., N.-C.L., H.-W.H., and C.-C.Y. wrote the manuscripts with input from all authors.