
Abstract
Background:
Late-onset glycogen storage disease type II is associated with variable muscle phenotypes. Epidemiological data suggest that its prevalence is lower in Belgium than in bordering countries like The Netherlands.
Objective:
We investigated whether such low estimated prevalence is due to missed diagnoses.
Methods:
We screened 100 patients with muscle phenotypes of undetermined origin using a dried blood spot test for alpha-acid glucosidase (GAA) activity. Patients with low activity at screening were re-tested by the same method and, if low activity was confirmed, GAA gene analysis was performed.
Results:
The screening test revealed lower than normal GAA activity in 15 patients, but in only two of them it was low enough to be considered in the disease range. Retesting confirmed lower than normal GAA activity in five patients, but in all of them it was above the disease range. A single patient carried a heterozygous known pathogenic GAA mutation, whose significance in this case remains undetermined.
Conclusions:
We conclude that reported low prevalence estimates in Belgium are not likely to be due to an underdiagnosis bias. Lower prevalence compared to neighbouring The Netherlands may be due to different ethnic stratification of our patients. Diagnostic strategies should keep into account the expected prevalence of a disease in specific populations.
ABBREVIATIONS LIST
INTRODUCTION
Late-onset type II glycogen storage disease type II (LO-GSDII) is a rare autosomal recessive metabolic muscle disease caused by deficiency of acid alpha-glucosidase (GAA). Its reported prevalence varies between 1:146,000 to 1:40,000 in different populations. The phenotype and the age of onset of LO-GSDII are widely variable, frequently leading to delayed diagnosis or misdiagnoses [1–3].
A dried blood spot test (DBS) for GAA activity has been recently developed and validated, demonstrating good reliability, high sensitivity and specificity [4–6].
In 16 recently conducted LO-GSDII screening studies, rates of detection varied between 0% and 4.6%. However, inclusion criteria, methodology and patients’ geographic origin differed [7–22] (see Table 1).
Findings of main recent studies concerning LO-GSDII screening
(1) with no cause nor association, (2) not precisely defined, (3) only one patient with two consecutive abnormal DBS was tested, (4) only this study was retrospective, other being prospective, (5) including 220 infantile onset suspected cases, no distinction was provided in the results between late-onset and infantile-onset cases. CK: creatine kinase, DBS: dried blood spot test, EMG: electromyography, LGMD: limb-girdle muscle dystrophy, LO-GSDII: late-onset glycogen storage disease type II, MRC: medical research council strength scoring, NA: not available.
Because enzyme replacement therapy (ERT) has proved its efficacy and safety in GSDII [23, 24] it is particularly important not to miss this diagnosis.
Epidemiological data suggest a lower prevalence of LO-GSDII in Belgium compared to The Netherlands. According to two independent sources, the Belgian Neuromuscular Registry recording patients in neuromuscular reference centers and the Sanofi Genzyme database recording ERT-treated patients, LO-GSDII prevalence seems particularly low in the French-speaking part of the country (Wallonia and Brussels), between 2.9 fold and 3.5 fold lower than in the Northern Flemish-speaking region (see Table 2). The highest current estimate of the number of LO-GSDII patients in Belgium is 45, based on which the prevalence in Belgium would be 45/11,209,044 = 3.49×10-6 compared to a five-fold higher prevalence of 17.54×10-6 in The Netherlands [2]. To assess whether such a low prevalence estimate may be due to missed diagnoses, we screened for LO-GSDII a series of patients with compatible muscle phenotypes who are followed in our neuromuscular clinic, which mainly serves Wallonia and Brussels.
Epidemiologic data concerning LO-GSDII in Belgium following different sources and compared to extrapolated in The Netherlands
* no data available for the NMRC UZ-Brussel/Inkendael and Liège-CHR La Citadelle. BNMDR: Belgian NeuroMuscular diseases Registry, GSDII: glycogen storage disease type II, LO-GSDII: late-onset GSDII.
MATERIALS AND METHODS
Design
We conducted an academic monocentric, prospective, interventional but non-therapeutic study. The study was sponsored by Sanofi Genzyme.
Inclusion and exclusion criteria
We included adult patients (>18 years old) with an unexplained muscle phenotype. The phenotype categories were stratified as: weakness, muscle pain, muscle fatigability, asymptomatic high CK level, acute rhabdomyolysis and muscle atrophy.
“Unexplained” indicates the lack of a diagnosis and the absence of clinical signs and ancillary investigations results suggesting secondary muscle involvement (e.g.: second motoneuron disease, peripheral neuropathy or central nervous system disorder). Exclusion criteria were age <18, absence of consent, previously performed GAA activity assay, other identified diagnosis explaining symptoms.
Clinical data collection
For all patients, the following data, when available/performed, were obtained from medical records: age at onset of symptoms or when an elevated CK level was first detected, age at sampling, sex, other diagnoses that had been considered or confirmed, highest CK level in the past medical history, and muscle biopsy findings.
Study procedures
After identification in the Erasme neuromuscular database, patients were recruited by phone. A blood sample was obtained for the DBS test and a creatine kinase (CK) assay. Patients were informed of a negative DBS test result by mail. When the test was positive or borderline, patients were invited to perform a second DBS test. A normal DBS re-test excluded LO-GSDII, whereas the diagnosis was not excluded when the DBS re-test was abnormal, in which case GAA gene analysis was performed. The complete flow-chart is shown in Fig. 1.
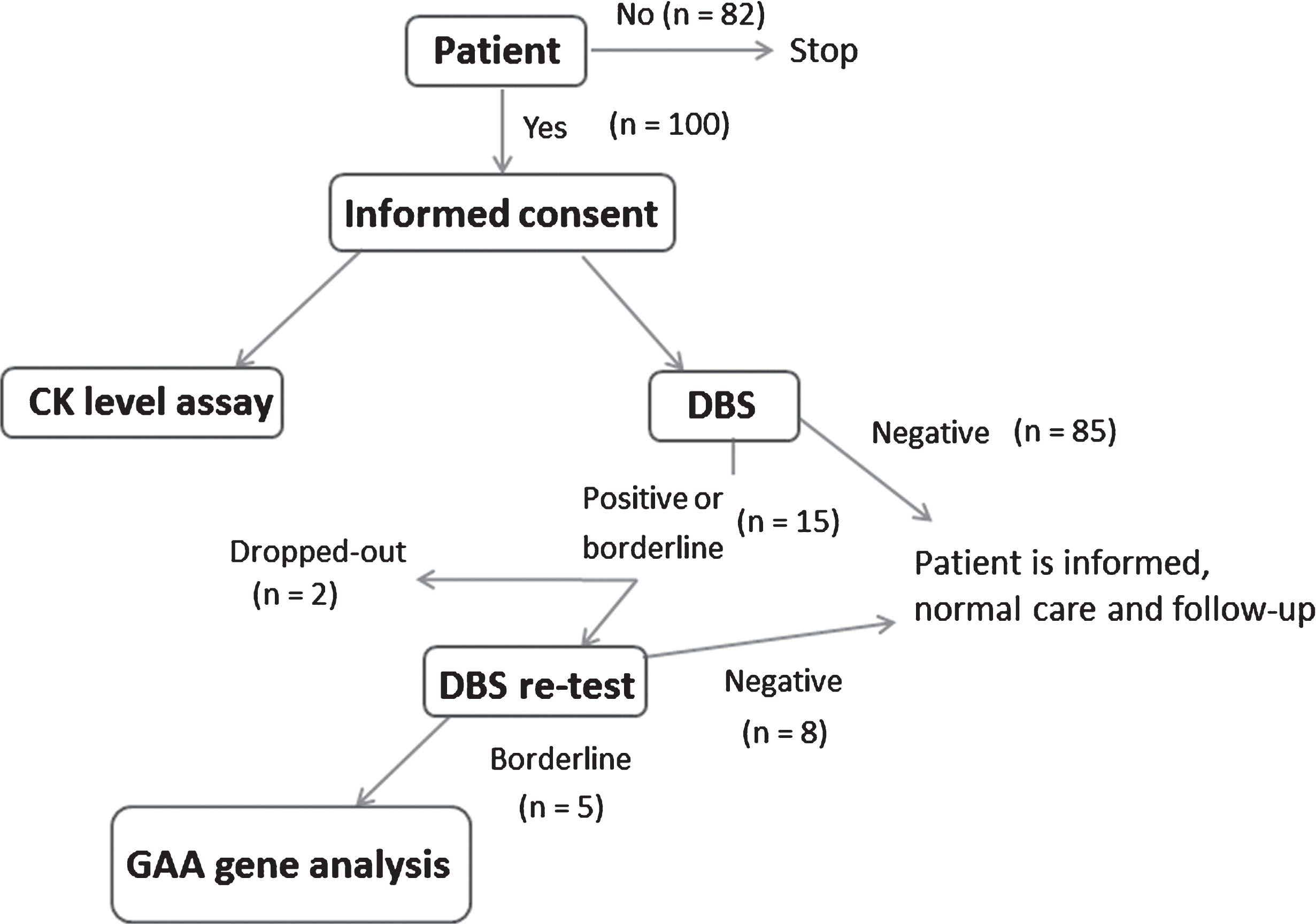
Chart flow concerning the recruitment of our patients. CK creatine kinase, DBS dried blood spot.
Sampling and enzyme assays
CK assay was performed in the laboratory of Erasme Hospital following the routine procedure. For the DBS test, blood was collected from a peripheral vein in an ethylenediaminetetraacetic acid (EDTA)-coated tube, a few drops were then spotted onto a filter paper, which was shipped to the Metabolic Diagnostics Unit, Hamburg University Medical Centre, Germany (Dr. Lukacs), where GAA activity was analyzed by fluorometric assay [7].
Gene sequencing
GAA gene analysis was performed if GAA activity by DBS test was below 0.9 nmol/spot×21 hours in 2 independently obtained samples. After deoxyribonucleic acid (DNA) extraction, all GAA coding exons and flanking intronic region were amplified by polymerase chain reaction (PCR) and sequenced. In addition, long-distance PCR was performed for deletion screening (ARCHIMED Life Science GmbH, Vienna, Austria; www.archimedlife.com).
Statistical analysis
Due to the small number of confirmed abnormal DBS test results, no formal statistical analysis was performed.
Ethics statement
Our research was conducted in accordance with the 1964 Helsinki Declaration, moreover, the protocol was approved by the local ethics committee of the Hôpital Erasme (Université Libre de Bruxelles, P2013/214) and written informed consent was obtained from all patients.
RESULTS
One hundred patients were screened between August 2013 and November 2015. Male/female ratio was 0.85. Mean age at sampling was 46 years old. Fifty-four had an elevated CK level. Patients were stratified according their clinical manifestations: weakness (37%), muscle pain (21%), muscle fatigability (20%), asymptomatic high CK level (14%), acute rhabdomyolysis (7%) and muscle atrophy (1%). A synthesis of epidemiological and clinical data is provided in Table 3.
Epidemiological and clinical data of the cohort
SD: sandard deviation, min: minimum, max: maximum, CK: creatine kinase.
Fifteen patients had low GAA activity at the first DBS screening, but only two were in the disease range (P7 and P13). Thirteen of them were re-tested, one withdrew consent and one was lost to follow-up. Low GAA activity was confirmed in 5 patients, although in none of them it was within the LO-GDSII range, defined as below 50% of the cut-off of 0.9 nmol/punch*21 h [25]. Genetic analysis of the GAA gene did not detect any potentially deleterious variant in 4 of these patients. One patient was heterozygous for the c.2238G < C variant, which leads to the aminoacid change p.Trp746Cys considered in the Pompe Rotterdam database as disease-causing, but potentially mild (Table 4).
Synthesis of the data concerning screened patients with first dried blood sport (DBS) positive or borderline
*Whithin the expected range for glycogen storage disease type II diagnosis. In bold, abnormal DBS confirmed.
DISCUSSION
We found no LO-GSDII cases in a series of 100 patients from Brussels and Wallonia with clinical features of adult-onset myopathy that may be compatible with this diagnosis, suggesting that the low estimated prevalence of LO-GSDII in this region is not due to an underdiagnosing bias. Such markedly lower prevalence compared to the Northern part of Belgium (Flanders) and neighboring The Netherlands therefore could reflect differences in the genetic makeup of the populations of these regions. However, it is possible that the prevalence of LO-GSDII in The Netherlands has been overestimated [2], particularly considering that the previously evoked existence of a founder effect [26, 27] is contradicted by several observations, such as the finding of numerous different mutations in Dutch patients and the lack of isolation of the Dutch population. In addition, patient recruitment in our neuromuscular reference center in Brussels, which serves the French-speaking population of Brussels and a large part of Wallonia, is likely to be less representative of the local population than in similar Flemish centers, because of recruitment bias. This is in part due to the longer distance to reach a university hospital in Wallonia than in Flanders, along with more diffuse socio-economic difficulties in Wallonia, as indicated by the lower income and higher rate of unemployment in Brussels-Wallonia than in Flanders. The consequent lower recruitment of patients from Wallonia, along with a highly heterogenous population, largely not of Belgian origin, living close to our university hospital made our sample very much ethnically mixed and not quite representative of the Belgian French-speaking population.
Previous similar studies (Table 1) showed different rates of detection of LO-GSDII, which may be due to the variability in inclusion criteria and in their definition. Not unexpectedly, it appears that when inclusion criteria are less strict and less rigorously defined, detection rate is lower. Compared to similar studies, we had a very low detection rate of LO-GSDII, but the highest rate of abnormal DBS screening tests. However, the definition of a positive DBS test was not homogenous among studies, making comparisons difficult. We chose to initially use relatively relaxed criteria to cast our net as wide as possible, but, different from some other studies, we required confirmation with a second DBS test before undertaking genetic analysis.
Our results confirm that the specificity of DBS is limited, as only 4/13 patients had a positive re-test and none had genetically proven LO-GSDII. Moreover, sometimes results of the first and the second test were highly divergent, as in the case of patient 7 who had a residual GAA activity of 34.4% in the first test and of 258.9% in the second test.
Some patients with an abnormal second DBS test but no detectable GAA mutation had a lower GAA residual activity than expected in heterozygous individuals (e.g. patient 8 compared to patient 13 in Table 4). The possible causes of low DBS in subject without GSDII include inappropriate sample collection and shipping, heterozygosity for GAA mutation or pseudodeficiency, mainly seen in Asian patients (p.Gly576Ser and p.Glu689Lys), in homozygosity, heterozygosity with or without a combination with a deleterious mutation [5, 16]. Inappropriate collection or shipping may be the cause of our 8 false positive results at initial testing. However, this is an unlikely explanation for the four patients who had a second abnormal DBS but carried no abnormal gene variants, including variants related to pseudodeficiency. In any case, their GAA activity, though lower than normal, was higher than usually found in GSDII patients. The only patient having a markedly low GAA activity was a heterozygous carrier of a disease-specific mutation. The observation that GAA activity determined by DBS test may be unspecifically low should remind the clinician to interpret with caution these results and seek further confirmation, either by enzyme activity assay in cells (fibroblasts, lymphocytes etc.) or by genetic analysis. One must remind, since we used GAA enzyme activity on DBS, that our study was not designed to assess GAA variant carrier frequencies. In consequences, the fact that only one out of the five genetically tested patients carried a pathogenic variant is not representative of the whole cohort.
We identified a heterozygous subject carrying the c.2238G>C (exon 10) GAA variant, labeled as potentially mildly pathogenic by the Erasmus Pompe Disease mutation database and reported in at least seven reports of Asian GSDII patients [28–34]. Data in the Exac® public database show a global allelic frequency of 2.99 10-4 and 0 counts in 10.306 alleles from African patients (our patient being from North Africa origin). We may speculate about a potential clinical impact of the heterozygous mutation or a potential modifying impact on other underlying neuromuscular etiologies. To the best of our knowledge, the question whether heterozygous patients may present an attenuated phenotype has never been addressed for GAA gene mutations. No conclusion can currently be drawn about our patient, who still has no alternative diagnosis and whose diagnostic workup, including muscle biopsy, is still incomplete.
Increased availability of next-generation sequencing will likely render screening tests like the DBS test obsolete, but the GAA enzymatic assay will continue to be used to confirm the pathogenicity of any new variant.
SOURCES OF SUPPORT
This study received financial support of Sanofi Genzyme.
CONFLICT OF INTEREST
Funding: This study was funded by Sanofi Genzyme (May 2013, no grant number provided by the Company). Author Gauthier Remiche
From 2008 to 2017, Dr Remiche received several advantages from Sanofi Genzyme, Santhera Pharmaceuticals, CSL Behring and C.A.F.-D.C.F. cvba-scrl concerning several activities such as: travel grants and financial support in order to attend scientific meetings, consultancy honorarium, speaker honorarium, grant to conduct academic study and grant to organize scientific meeting. Author Zoltan Lukacs Received research and travel grants from Genzyme B.V. (NL), and BioMarin Ltd (UK). Author David C. Kasper declares that he/she has no conflict of interest. Author Marc Abramowicz declares that he has no conflict of interest. Author Massimo Pandolfo Dr. Pandolfo has received personal compensation for consulting, serving on a scientific advisory board, speaking, or other activities with Voyager Therapeutics, Biomarin, Apopharma. Dr. Pandolfo has received personal compensation in an editorial capacity for Neurology Genetics. Dr. Pandolfo has received royalty, license fees, or contractual rights payments from Athena Diagnostics. Dr. Pandolfo has received research support from Biomarin, Voyager Therapeutics.
*Research involving human participants and/or animals
Ethical approval: All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. CE Code (Hôpital Erasme, Université Libre de Bruxelles, 808 route de Lennik, 1070 Brussels, OM021): P2013/214. EudraCT/CCB: B406201317951, 16/07/2013.
*Informed consent
Informed consent was obtained from all individual participants included in the study.
Footnotes
ACKNOWLEDGMENTS
We acknowledge Mr Nick Alaerts and Mr Alessandro Fadel for their precious support to collect samples and patients for their kind collaboration. We also kindly acknowledge Dr Corinne Bleyenheuft from Belgian Neuromuscular Disease Registry (BNMDR) - Belgian Scientific Institute of Public Health for providing epidemiologic data.