
Abstract
Background:
Universal spinal muscular atrophy (SMA) newborn screening was implemented in California on June 24, 2020.
Objective:
We describe California’s experience with the first 18 months of SMA newborn screening, including our assay methodology, timeliness of screening and follow-up milestones, and clinical and epidemiological outcomes observed.
Methods:
Dried blood spots are screened for SMA using multiplex real time polymerase chain reaction (RT-PCR) to detect deletions of exon 7 in the survival of motor neuron 1 (SMN1) gene. Short-term follow-up data is collected from clinical staff via an online data collection tool.
Results:
In the first 18 months, 628,791 newborns from California’s diverse population were tested for SMA. Thirty-four screened positive and were confirmed to have the disorder. Infants were referred, diagnosed, and treated at a median of 8, 12, and 33 days of life, respectively. Nearly all infants received the desired treatment modality, and 62% received treatment while still asymptomatic.
Conclusions:
SMA newborn screening is a highly sensitive and specific test which identifies infants with SMA early when treatment is most effective. Even with newborn screening’s success in facilitating early intervention, there is still work to be done to expedite treatment, especially for infants with the most severe form of the disease.
INTRODUCTION
Spinal muscular atrophy (SMA, OMIM *600354) is an autosomal recessive neuromuscular disorder and has been reported as the leading genetic cause of infant mortality. For 95-98% of cases, SMA is caused by a homozygous deletion of the exon 7 region on the survival of motor neuron 1 (SMN1) gene, which codes for the spinal motor neuron (SMN) protein. Untreated, this protein deficiency leads to degeneration of spinal cord motor neurons causing a multi-system condition associated with progressive dysfunction of musculature within the body and signs and symptoms which include hypotonia, dysphagia, limb weakness, contractures, paralysis, and often respiratory failure [1–5].
The severity of the condition is partially modified by the copy numbers of survival of motor neuron 2 (SMN2) gene, a paralog of SMN1. Infants with more copies (4 or more copy number) of the SMN2 gene generally have a milder phenotype and may not present clinically until later in childhood or adulthood. They may experience muscle weakness and other relatively mild symptoms but are able to have a normal lifespan. Those with fewer copies (0-3 copy number) of the SMN2 gene have an earlier onset of disease, a more acute clinical presentation, and a more severe prognosis. If left untreated, newborns with the more severe forms often die before two years of age from respiratory failure [1, 4].
Diagnosis of SMA involves DNA testing for the absence of exon 7 on the SMN1 gene and the copy number of the SMN2 gene. To better determine disease progression, physicians use a combination of neuromuscular physical exams, laboratory and clinical tests, and neurodevelopmental assessments such as CHOP INTEND and HINE [6–8].
With the advent of cutting-edge therapies, including nusinersen, onasemnogene abeparvovec, and risdiplam, the natural progression of SMA has been altered [7, 10]. In 2016, nusinersen became the first drug to gain US Food and Drug Administration (FDA) approval to treat SMA. It is intrathecally injected into the patient a few times a year, and works by integrating exon 7 into the SMN2 mRNA, thereby increasing SMN protein production. In 2019, a gene-replacement therapy, onasemnogene abeparvovec, gained FDA approval. It is a one-time intravenous infusion in which AAV9 viral vectors deliver the SMN1 transgene to motor neurons, increasing production of SMN proteins. Risdiplam most recently had FDA approval expanded for treatment in the newborn population in 2022. It is orally administered and increases SMN protein production by modifying splicing of the SMN2 mRNA to integrate exon 7 [10]. With these new therapies, newborns treated within days or weeks after birth appear to retain neuromuscular function and therefore do not experience the fatal consequences they once would have.
Newborn screening is poised to quickly identify newborns who have SMA and help provide early access to lifesaving, and disability-modifying treatments. Massachusetts and Utah were the first states in the US to offer statewide newborn screening for SMA in 2018 [11]. On July 2, 2018, the Secretary of Health and Human Services signed SMA onto the Recommended Uniform Screening Panel (RUSP) [12, 13]. California is legislatively mandated to expand statewide screening of newborns for any disease detectable in blood samples no later than two years after it is adopted by the RUSP [14] and consequently added SMA to the screening panel on June 24, 2020. As of August 29, 2022, forty-seven states universally screened newborns for SMA, accounting for 97% of babies born in the US [15, 16].
In this paper, we describe California’s experience with the first 18 months of SMA newborn screening, including our assay methodology, timeliness of screening and follow-up milestones, as well as clinical and epidemiological outcomes observed in short-term follow-up. With more than 400,000 babies born each year amidst a racially and ethnically diverse population, this study provides a unique real-world description of the efficacy of newborn screening for SMA.
MATERIALS AND METHODS
Assay methodology
In California, dried blood spots (DBS) are collected from 12 through 48 hours after birth for 98% of hospital born infants, excluding the few whose parents opt out for religious reasons. Specimens for SMA screening are sent to the California Genetic Disease Screening Program (GDSP) in Richmond, CA, where specimens are processed Monday through Friday. California utilizes the CDC-developed multiplex real time polymerase chain reaction (RT-PCR) method to detect deletions of exon 7 in the SMN1 gene [17, 18], which does not detect heterozygotes. As a quality control process, all screen-positive specimens are analyzed by QX200™ Droplet Digital PCR™ (ddPCR) (BioRad, Hercules, CA) to quantify the copy numbers of SMN1 and SMN2, using the same DNA extract as the one used for the RT-PCR analysis. Only SMN1 results are reported to providers.
Three millimeter punches are directly punched into 96-well plates using the DBS Puncher Instrument® (Perkin Elmer, Waltham, MA). In order to provide genetic screening on up to 2,500 specimens within an eight-hour workday, we operate two automated high precision Microlab STARPLUS® (Hamilton Company, Reno, NV) liquid handlers with the Extracta DBS® extraction buffer (Quantabio) and four QuantStudio™ 7 Flex Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA) instruments processing 384-well plate format. The liquid handler on-deck instruments include plate peeler, plate sealer, centrifuge, shaker heating and cooling blocks to enable automation of the workflow from DNA extraction at 95°C, storage of DNA extracts and RT-PCR reagents at 4°C, and dispensation of samples and reagents into 384-well plates ready for RT-PCR analysis. Four 96-well plates with specimens can be processed within three hours by the liquid handlers, and each RT-PCR analysis of a 384-well plate can be completed within two hours.
The DNA extracts are aliquoted into 384-well plates (ThermoFisher Scientific, Waltham, MA) and mixed with custom PerfeCTa Multiplex qPCR ToughMix (Quantabio, Beverly, MA). The 384-well plates are transferred to the RT-PCR instruments for fluorescence quantification of reference gene Ribonuclease P Subunit p30 (RPP30) with the HEX fluor, SMN1 with the Cy5 fluor, and T-cell receptor excision circles (TREC) with the FAM fluor. Primers and probes are purchased from Integrated DNA Technologies (IDT, Newark, NJ). A Locked Nucleic Acid (LNA) probe is designed to target SMN1 in the exon 7 region with high specificity. Fluorescent signals for RPP30 of cycle threshold (Ct)≤28 and SMN1 Ct≥30 in the RT-PCR assay indicates a screen positive for SMA. A screen-negative specimen has fluorescent signals for RPP30 of Ct≤28 and SMN1 of Ct < 30. A specimen with incomplete results has a fluorescent signal for RPP30 of Ct≥28. Both screen-positive and incomplete specimens are retested in triplicates on three fresh punches the next business day.
Referral algorithm and clinical follow-up
SMA results usually post one business day after they are determined, at which point cases are assigned to area service centers who then notify primary care physicians and coordinate a referral. Newborns who screen positive for SMA are referred to specialists at one of fourteen neuromuscular special care centers (SCCs) across California that provide genetic counseling, order confirmatory testing, determine a case resolution, and coordinate treatment, if necessary. During the study period, GDSP paid for multiplex PCR/CE testing (AmplideX® PCR/CE SMN1/2 Plus Kit, Asuragen, Austin, TX) at a contracted laboratory to confirm the absence of the SMN1 gene and to determine SMN2 copy number, which is performed on a new blood specimen. All other follow-up testing and treatments are covered by the family or a third-party payer.
Contracted SCCs report to GDSP on every referred case via an online short-term follow-up (STFU) data collection tool in our Screening Information System (SIS), which includes information about services provided to the newborn from birth through the initial diagnostic work-up and initiation of treatment. Additionally, SCCs use a similar long-term follow-up (LTFU) data collection tool in SIS to provide an annual report for five years containing updated health outcome data on every child diagnosed with SMA that continues to be followed by an SCC [19].
Treatment barriers and delays assessment
One year into screening, we surveyed the SCCs for each of 24 newborns that screened positive for SMA from June 24, 2020 through June 23, 2021 to gather additional information about treatment delays and barriers. Eight questions assessed the clinicians’ opinions on whether the patients received the preferred treatments, whether the treatments were delivered in a timely manner, and the factors that may have contributed to treatment choices or delays (see Supplementary Document). For cases where a survey was not completed, we searched case notes entered by SCC staff into the SIS for mention of delays, barriers, elevated adeno-associated virus serotype 9 (AAV9) antibody titers, or insurance authorization issues.
Study population and statistical analysis
For this report, we retrospectively reviewed California newborn screening and follow-up data for specimens accessioned from June 24, 2020 through December 23, 2021. Race/ethnicity data was self-reported on the NBS test request form as per California regulations. When multiple race/ethnicities were selected they were assigned a single race/ethnicity using the following priority: Native American, Black, Hispanic, Asian, then White. A prior known family history of SMA homozygosity or heterozygosity was recorded for infants who were screen positive for SMA if specialists answered “Yes” to “Was disorder diagnosed prenatally?”, or if case notes mentioned the child having a sibling with SMA, or if case notes mentioned the child having at least one parent that was a known heterozygote.
Descriptive statistics and confidence intervals were derived using Microsoft Excel®. Wilcoxon rank-sum tests and Fischer’s exact tests were performed using SAS software, Version 9.4 of the SAS System for Windows (copyright © 2002–2012 SAS Institute Inc.; SAS and all other SAS Institute Inc. product or service names are registered ™s or ™s of SAS Institute Inc., Cary, NC, USA).
RESULTS
In the first 18 months, 628,791 newborns were screened for SMA, and 34 screened positive. Negative results posted in SIS at a median of 5 days of life, and positive screening results posted at a median of 7 days of life (Fig. 1), a median of 2 days later than negatives due to retesting of positive specimens.
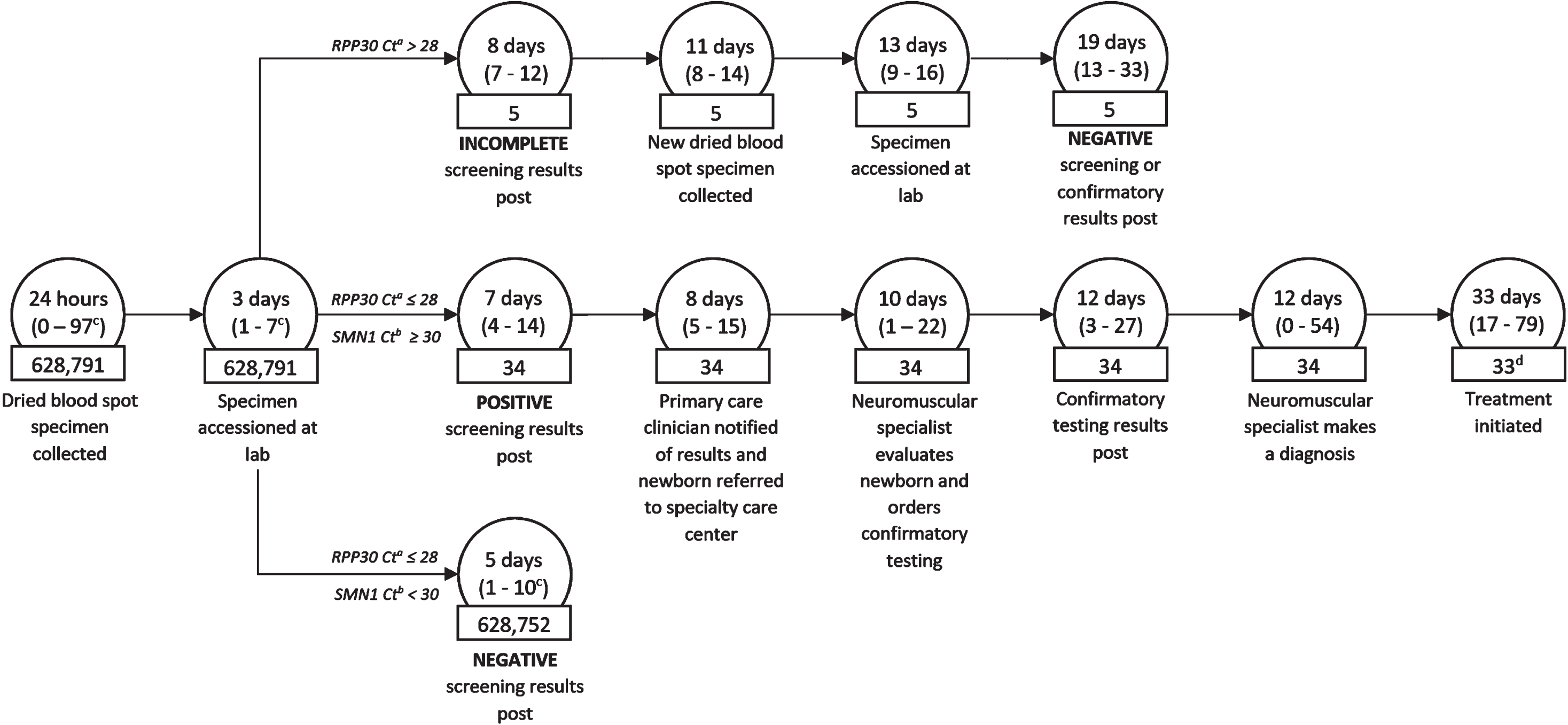
The SMA newborn screening and follow-up process in California. Circles contain the median (and range) time from birth. Rectangles contain the number of newborns at each step of the process which was observed in the first 18 months of SMA newborn screening. (a) RPP30 Ct is the ribonuclease P protein subunit p30 gene (reference gene) cycle threshold (b) SMN1 Ct is the survival motor neuron 1 gene cycle threshold (c) Represents the range up to 99th percentile. Due to extraordinary circumstances, some clients had specimen collections as late as 20,175 hours, specimens accessioned as late as 842 days, and negative results as late as 845 days. (d) One infant expired before treatment was initiated.
Five newborns screened had initial incomplete results and had dried blood spots recollected at a median of 11 days of life. Screening results were inconclusive for two infants due to Ct slightly above the cutoff of 28 for RPP30 in both the initial and repeated DBS specimens. Those two cases were subsequently referred to the SCC as incomplete. A new blood sample was collected for each of the infants, and results were obtained through our contracted confirmatory laboratory. All five infants with initial incomplete screening results were either found to be screen negative or confirmed to have 2 copies of the SMN1 gene by a median of 19 days of life.
All 34 newborns with a screen-positive result were confirmed to have SMA (Table 1), representing a birth prevalence of 1 in 18,494 newborns (95% confidence interval [CI]: 1 in 13,841 to 1 in 27,858). Prior family history was reported in 13 cases (38%), and one additional case was reported as missed prenatally due to the father being a suspected silent heterozygote, carrying both copies of SMN1 on a single allele. No false negatives from newborn screening have been reported.
Birth prevalence rates for infants diagnosed with SMA by SMN2 copy number, sex, and race/ethnicity
The GDSP contract laboratory was utilized to confirm 56% of cases (19/34), though results were reported for every case. Confirmatory testing revealed that 47% of newborns (16/34) had 2 copies of the SMN2 modifier gene, 35% (12/34) had 3 copies, and 18% (6/34) had 4 or more copies. No infants with 0 or 1 copy number for SMN2 were identified through our newborn screening. Confirmatory results were reported to providers at a median of 12 days of life, and infants were diagnosed at a median of 12 days of life.
All but one of the 34 infants had clinical assessment data reported during the short-term follow-up period, with the last reported clinical assessment occurring at a median of 16 days (range 4-147 days). Twelve infants (36%) were reported to have clinical findings related to the SMA diagnosis during the STFU period, including 50% (8/16) of infants with 2 copies of the SMN2 gene compared to 25% (3/12) of infants with 3 copies and 20% (1/5) with 4 copies. The most common clinical finding was hypotonia/poor muscle tone, observed in 8 infants (Fig. 2). Seven infants were reported to have abnormal assessments or procedures, including four with abnormal CHOP INTEND assessments, and 1 infant each with an abnormal HINE assessment, echocardiogram, electrocardiogram, and neurological exam, as determined by the infants’ clinician.
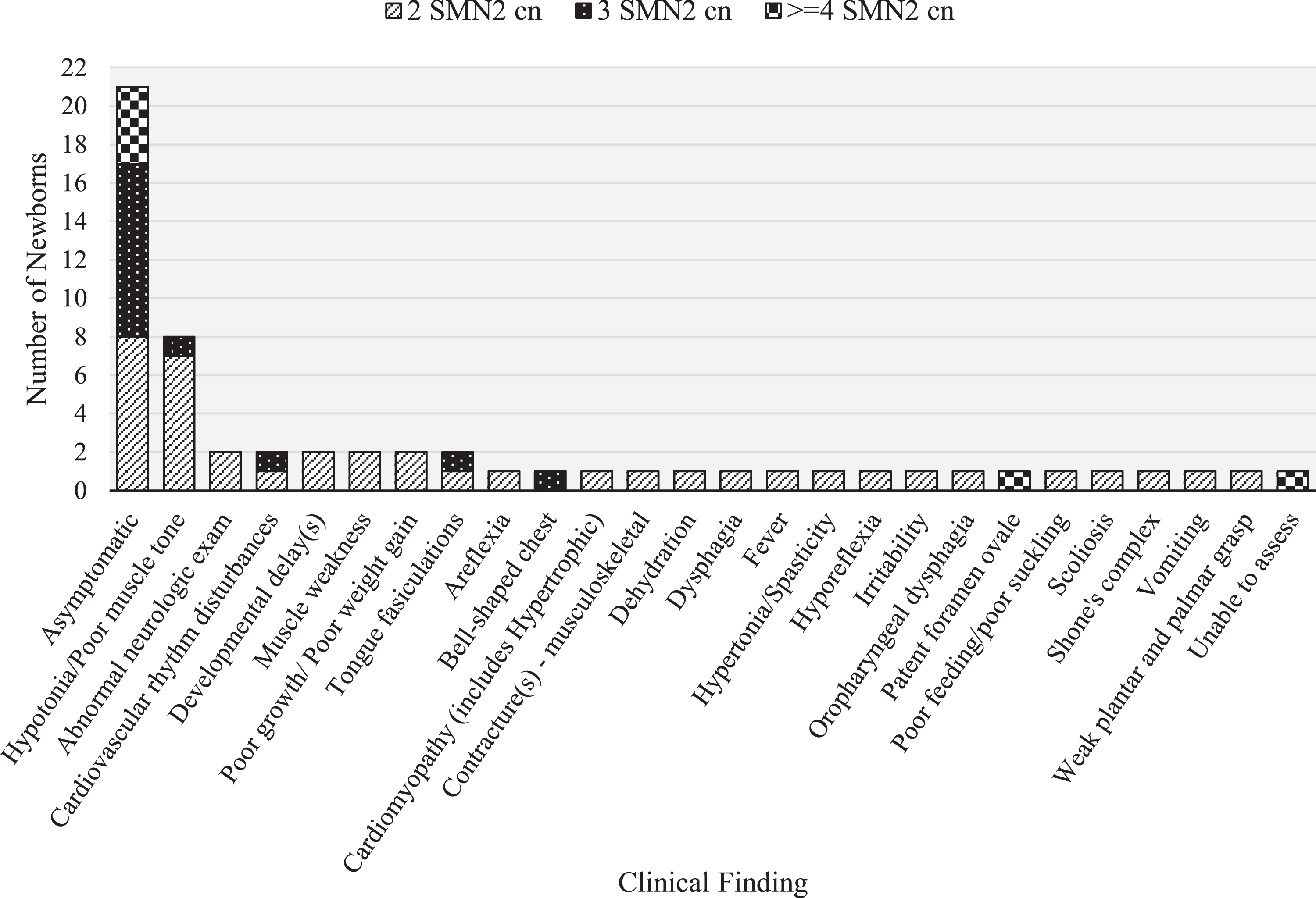
Clinical findings observed in 34 newborns confirmed with SMA, with fill patterns denoting SMN2 copy number (cn). Some newborns had more than one clinical finding reported.
Treatment was administered to 33 infants at a median of 33 days of life, with 29 receiving onasemnogene abeparvovec, 3 receiving nusinersen, and 1 receiving both onasemnogene abeparvovec and nusinersen (Table 2). One infant with 2 copies of the SMN2 gene expired prior to treatment due to complications of a congenital heart defect. Treatments were administered sooner, median of 30 days of life, for infants with 2 or 3 copies of the SMN2 gene, compared to infants with 4 or more copies of the SMN2 gene, who received treatment at a median of 57 days of life (Wilcoxon Scores, Z = 2.34, p < 0.01).
Treatment type and follow-up milestones for 34 newborns confirmed with SMA
Twenty-four surveys, one for each SMA case confirmed in the first year of screening, were sent to clinicians at 11 neuromuscular centers to assess delays and barriers to treatment. Seven centers responded, completing surveys for 18 of the 24 confirmed cases. All but two (16/18) infants were administered the preferred treatment modality. One infant had elevated AAV9 antibody titers, preventing the infant from receiving the preferred treatment modality, onasemnogene abeparvovec. The other had medical care complications, which required that the patient be stabilized with nusinersen before the preferred onasemnogene abeparvovec treatment could be administered. An additional 5 infants for whom we received no survey were identified to have elevated AAV9 antibody titers based on a review of case notes, including 2 who were administered nusinersen.
Half (9/18) of infants were reported to have had treatment administered in a timely manner. This trend was consistent for infants covered by private insurance (4/8) as well as publicly provided insurance (5/10). The most common barriers, or reasons for delay to treatment were problems with insurance authorization (n = 6), logistical issues getting the patient into the center for treatment (n = 2), delays due to SMA-related health issues (n = 1) or other health issues (n = 2), delays in receiving confirmatory results (n = 2), and delays at the pharmacy (n = 1). For 2 cases, clinicians expressed that delays may have been compounded by the fact that they were new to the newborn screening process.
Of the 34 children diagnosed with SMA, 22 were at least one year old at the time of this study and in active follow-up, making them eligible for a LTFU report. We received LTFU reports for 16 (8 with 2 SMN2 cn, 7 with 3 cn, and 1 with> =4 cn). Six were reported to have symptoms related to the disorder, including 3 that were reported to be asymptomatic in the short-term follow-up period. All 6 symptomatic children had 2 copies of the SMN2 gene. Three of the symptomatic children were reported to have delays or barriers to treatment.
DISCUSSION
In the first 18 months of SMA newborn screening in California, we observed an SMA birth prevalence of 1 in 18,494. This observation is lower than what was expected based on homozygote frequencies in the literature of 1 in 10,000 [5], but within the range reported by other state and international newborn screening programs [11, 20–22]. Lower than expected incidence rates may be due to a couple of reasons. First, previous estimates in the literature were based on predictions from homozygote frequencies and cases that presented clinically, whereas newborn screening is an essentially population-based assessment. Second, awareness and uptake of prenatal heterozygote screening may have increased since incidence estimates were published.
Previous studies suggest that there are differences in heterozygote frequencies between races and ethnicities [23, 24], however, any differences in incidence rates that we observed between race and ethnic groups were not statistically significant due to the small sample sizes underlying the incidence rates (Fischer’s Exact, p > 0.1). We may also have some reporting bias, as race/ethnicity is supposed to be self-reported but is often selected by hospital staff completing the test request form based on what is known about the mother and may not be accurate.
SMA prenatal heterozygote screening is currently recommended for all women who are pregnant or considering pregnancy [25, 26], however we only observed that 38% (13/34) of newborns had a prior known risk for SMA before birth. While data is limited on rates of heterozygote screening, other newborn screening programs have reported that 43-56% of screen-positive newborns had at least one parent that was a known heterozygote [11, 20]. Our lower rate might reflect differences in prenatal care practices, or they may be due to limitations in our data collection. Our STFU form asks specialists, “Was disorder diagnosed prenatally?”, which may not elicit an affirmative answer when the parental heterozygote status was known but the fetus was not diagnosed. To supplement these data, we also queried case notes to find newborns with prenatal heterozygote screening or a sibling who was diagnosed with SMA. Regardless, we may have an underestimation of cases that were either diagnosed prenatally or that had a prior family history.
No false negatives have been reported at the time of this study. A limitation of our RT-PCR methodology is that it will miss around 4% of newborns who are compound heterozygotes for SMN1 variants or who have a variant on another part of the gene [23]. While these cases fall outside the target of newborn screening, we do ask that clinicians notify us of these rare occurrences, and none have been reported yet. Since clinical diagnosis can be delayed beyond one year [27], missed cases or cases outside the target of screening might be reported as time progresses.
Five newborns had initial incomplete results, including 2 whose reference gene Ct remained above the incomplete cutoff. This finding has prompted us to investigate the cause of the incomplete results. The borderline reference gene cycle thresholds could be due to the existence of RPP30 sequence variants in the California population, resulting in the RT-PCR primers not optimally detecting the reference gene DNA sequences for some individuals. A cutoff change may be warranted. However, we have not been able to rule out presence of something in the DBS specifically inhibiting reference gene extension or probe binding to the PCR product.
Despite the fact that we obtain SMN2 copy number through our internal ddPCR quality control step, we chose to contract with a separate laboratory to provide SMN2 copy number to the SCCs. As a result, newborns are referred before SMN2 copy number is known, and SMN2 copy number results have been available to specialists at a median of 12 days of life. Conversely, results from our internal ddPCR analysis yields results when newborns are referred for follow-up, at a median of 8 days of life. Still, advantages to contracting out this work remain. Specialists will still need to collect another sample to confirm the absence of the SMN1 gene in their patient, and in most instances, to assess AAV9 titer levels. Both of these tests are often offered as a panel with SMN2 copy number testing and are generally necessary before treatment can begin. Our laboratory is working toward validating our ddPCR testing and thus far has found concordance between our ddPCR results and the third-party confirmatory laboratories for all 34 screen-positive samples. As a result, considerations are being made to report SMN2 copy number with positive newborn screening results.
In our population, newborns who screened positive for SMA and found to have 2 or 3 copies of the SMN2 gene were treated at a median of 30 days of life. This aligns with timelines from other NBS programs where newborns were treated at between 24 and 36 median days of life [20–22]. It is also well below the median age of symptom onset for clinically diagnosed cases (2.5, 8.3, and 39.0 months for SMA type I, II, and III respectively) [27], highlighting the value of early detection through newborn screening.
By surveying neuromuscular clinicians, we found that insurance authorization and health status of the infant created barriers to preferred treatment options and contributed to delays in treatment administration. There is also some indication that delays were related to the newness of the newborn screening process for SMA, and overtime, we would expect these types of delays to be minimized as SMA screening becomes firmly established. Still, we were able to identify several areas where our newborn screening program could address delays, which includes working with our colleagues in other departments to streamline the insurance approval process, advocating for quicker confirmatory test turn-around times, and helping neuromuscular centers navigate the newborn screening process.
We found that 36% of confirmed newborns showed symptoms of SMA in the short-term follow-up period, with rates higher in those with 2 copies of the SMN2 gene. In addition, all of the children who were symptomatic at their 1-year follow-up had 2 copies of the SMN2 gene and half of them experienced delays or barriers to treatment. This underscores the importance of identifying SMN2 copy number as soon as possible and expediting treatment, especially for newborns that are more likely to have more severe clinical outcomes.
In the coming years newborn screening programs must monitor long-term follow-up data to fully evaluate the effectiveness of SMA newborn screening. Moreover, as children are being treated asymptomatically, it will become more important to monitor outcomes with standard periodic clinical assessments which may go beyond motor milestones that were the historical standard.
Conclusion
In California’s first 18 months of experience screening over 600,000 newborns for SMA, 34 newborns were successfully identified without any false negative or false positive screening results. Access to preferred treatment modalities was demonstrated and time to treatment was dramatically improved compared to clinically diagnosed cases. The median time to treatment in the California program was comparable to other SMA newborn screening programs and areas to improve early intervention have been identified. This will require improved coordination between the newborn screening program, special care centers, third-party payers, confirmatory laboratories, and pharmaceutical companies. Taken together, the efficacy of newborn screening for SMA has been demonstrated. Newborns were successfully identified shortly after birth allowing for critical and timely disease-modifying treatment.
Footnotes
ACKNOWLEDGMENTS
We want to extend our thanks to the staff at the state-contracted California Neuromuscular Special Care Centers who provide follow-up diagnostic services for referred newborns and data for our ongoing program evaluation. Additionally, we are grateful to our Area Service Center staff for the work they do to ensure smooth and timely referrals for our NBS families. We would also like to thank Dr. Francis Lee and his group at the CDC for sharing their expertise. Finally, SMA newborn screening would not be possible without our colleagues involved in the original implementation, testing and day-to-day operations, who include Lifan Shih, Lawson Wu, Preeti Bhattacharjee, Lauren Tom, Sergio Diaz, Jeffrey Kurosaka, Jordan Velasquez, Robin Cooley, and Greta Nash.
CONFLICTS OF INTEREST
The authors have no conflict of interest to report.
ETHICS DECLARATION
The California Health and Human Services Agency’s Committee for the Protection of Human Subjects has determined that the program evaluation and surveillance activities conducted by the California Newborn Screening Program is exempt research per federal guidelines, section 46.101(b)(4)(ii)). All of the work presented in this manuscript was carried out as program evaluation and surveillance activities of the California Newborn Screening Program and therefore falls under federal guidelines section 46.101(b)(4)(ii).