
Abstract
Background:
Myotonic dystrophy type 2 (DM2) is a multisystem disorder, mostly presented with mild but heterogeneous spectrum of symptoms.
Objective:
The aim of this research was to provide detailed sociodemographic, clinical and laboratory data of a large DM2 cohort from the Serbian registry.
Methods:
In 2008, we started to prospectively enter data of all DM patients. We also retrospectively collected data of patients hospitalized from 1990 until 2008.
Results:
At the end of 2017, registry comprised 87 (68%) of 128 genetically confirmed DM2 patients in Serbia, i.e. 1.2 registered cases per 100,000 inhabitants. Female subjects were more prevalent (63%). The diagnostic delay was 11.8±11.3 years. The most common first symptoms in our patients were lower limb weakness, handgrip myotonia and limb pain, although some percentage of patients presented with cataracts or extrapyramidal symptoms and signs. Lens opacities were present in 75% of patients. Severe ECG abnormalities were noted in 8% and pacemaker was implanted in 5% of DM2 subjects. Pulmonary restriction was observed in 10% of DM2 patients. Insulin resistance and diabetes mellitus were frequent in our cohort (21% and 17%, respectively). Male subjects more frequently had snoring, baldness, sterility, polyneuropathy, lower HDL and higher glycaemia, while waddling gait and increased muscle reflexes were more common in females.
Conclusions:
This registry offers a spectrum of different features presented in Serbian DM2 population, which could be at service of earlier diagnosis and better treatment.
INTRODUCTION
Myotonic dystrophy type 2 (DM2) is a multisystem disorder, inherited in an autosomal dominant pattern, and characterized by slowly progressive proximal muscle weakness, myotonia, muscle pain and cataracts [1, 2]. DM2 is caused by CCTG repeat expansion in intron 1 of the CNBP (cellular nucleic acid-binding protein) gene [3]. Although clinical presentation of DM2 is very similar to myotonic dystrophy type 1 (DM1), it also has some major distinctions. In contrast to DM1, DM2 has later disease onset, most of the symptoms seem to be milder and with better prognosis [3]. Information about DM2 is still insufficient and diagnostic delay for DM2 is above 10 years [4]. By creating the Serbian DM2 registry, our aim was to collect data on the epidemiological and clinical characteristics of DM2 with intention to contribute data to the TREAT-NMD global DM registry, too [5]. Secondarily, we sought to promote clinical research in this field. The clinical data collected in the Registry can also be used to stratify for inclusion criteria in potential clinical trials of new therapeutic strategies.
The aim of this research was to provide detailed sociodemographic, clinical and laboratory data of a large DM2 cohort from the Serbian registry. We also aimed to assess gender differences and to look for the influence of disease duration on different symptoms and signs.
MATERIALS AND METHODS
Serbian DM Registry was named Akhenaten after the pharaoh of Egypt who might suffered from this disease according to some sources [6]. It was founded in 2008 at the Neurology Clinic, Clinical Centre of Serbia, School of Medicine, University of Belgrade. Formation of the registries for rare neuromuscular disorders including myotonic dystrophies was approved by the Ethic Committee of the School of Medicine University of Belgrade. All patients gave informed consent to share their data in the Registry and to use their anonymous data in scientific publications.
In 2008, we started to prospectively enter data of all DM patients visiting the Neurology Clinic. We also retrospectively collected data of all patients hospitalized at our Clinic from 1990 until 2008. All previously diagnosed patients were contacted and reexamined, except three patients who passed away. All data comprised in Akhenaten are based on the questionnaire completed by a neuromuscular specialist. From 2008 onward, neuromuscular specialists from all around Serbia, Montenegro and the Republic of Srpska (Bosnia and Herzegovina) sent their patients to our referral center.
In all of our DM2 patients clinical and electrophysiological diagnosis was genetically confirmed by repeat primed polymerase chain reaction (RP-PCR) showing the presence of increased number of CCTG repeats in the CNBP gene [7]. This method was introduced in 2013 in the laboratory at the Center for Human Molecular Genetics, University of Belgrade. This is the only laboratory in Serbia, Republic of Srpska and Montenegro that performs DM2 diagnosis. Different sociodemographic, clinical and laboratory features entered in the Registry were analyzed in our research. Presence of ptosis, mastication and facial weakness, swallowing difficulties, sternocleidomastoid and trapezius muscle weakness, presence and duration of active/percussion hand myotonia, as well as electromyographic data represented a wide range of muscular symptoms which were included. Concentric needle electromyography was performed in deltoid, biceps, extensor digitorum communis, flexor digitorum profundus, vastus lateralis, tibialis anterior, and thoracic paraspinal muscles. Ten needle insertions per muscle were made in order to find myotonia, including classic waxing-waning or less specific waning discharges. To evaluate the muscle strength in limbs of our patients we used the Medical Research Council (MRC) 0–5 point scale (0 - no movement, 5 - normal strength) [8]. The strength of the weakest muscle group of proximal upper limbs, proximal lower limbs, distal upper limbs and distal lower limbs was added, where the maximum score was 20 [9]. Ophthalmologic examination included presence of cataract, eye pressure measure and fundus findings. Cardiologic evaluation comprised blood pressure measurement, electrocardiography (ECG) and echocardiography analysis, and presence of pacemaker. Severe ECG abnormality was defined according to Groh’s definition given for DM1 patients: rhythm other than sinus, PR interval of 240 ms or more, QRS duration of 120 ms or more, second degree or third-degree AV block [10]. Left ventricular ejection fraction (EF) was also noted during cardiologic examination, and it was considered pathological when below 55%. Pulmonary restriction was defined as forced vital capacity (FVC) < 90% of expected. Detailed laboratory examination included: glycaemia and serum insulin levels, HOMA index of insulin resistance (serum glucose x serum insulin/22.5), oral glucose tolerance test (after 120 minutes), serum triglycerides, cholesterol, HDL and LDL levels, thyroid function, and serum electrolytes levels.
We used Kolmogorov-Smirnov to assess normality of data. Methods of descriptive statistics were used, including proportion, mean and standard deviation. For comparison between groups, chi square test, Mann-Whitney U test and Student t test were used, as appropriate. Spearman’s rho was used for correlations. Level of statistical significance was 0.05.
RESULTS
The Akhenaten Registry has comprised data on 89 DM2 patients so far which is 69.5% of all DM2 patients genetically diagnosed in Serbia in the only laboratory that performs this analysis. Two patients were excluded from the analysis in this study since they have genetically confirmed both DM1 and DM2 (these patients have been previously published in reference [11]). As expected, the highest number of registered DM2 patients is observed in the district around the largest neuromuscular center in Belgrade, while there were no registered patients in nine of 25 regions of Serbia (Fig. 1). Number of registered patients in Serbia overall was 1.2 per 100 000 inhabitants with the highest regional prevalence of 2.4 registered cases per 100 000. From 2008–2013, a total of twenty patients have been included in the Registry. Maximal number of 21 DM2 patients was registered in 2013 when molecular-genetic analysis for DM2 was introduced in Serbia. From 2013 onward, a steady number of 11–12 patients per year was registered.
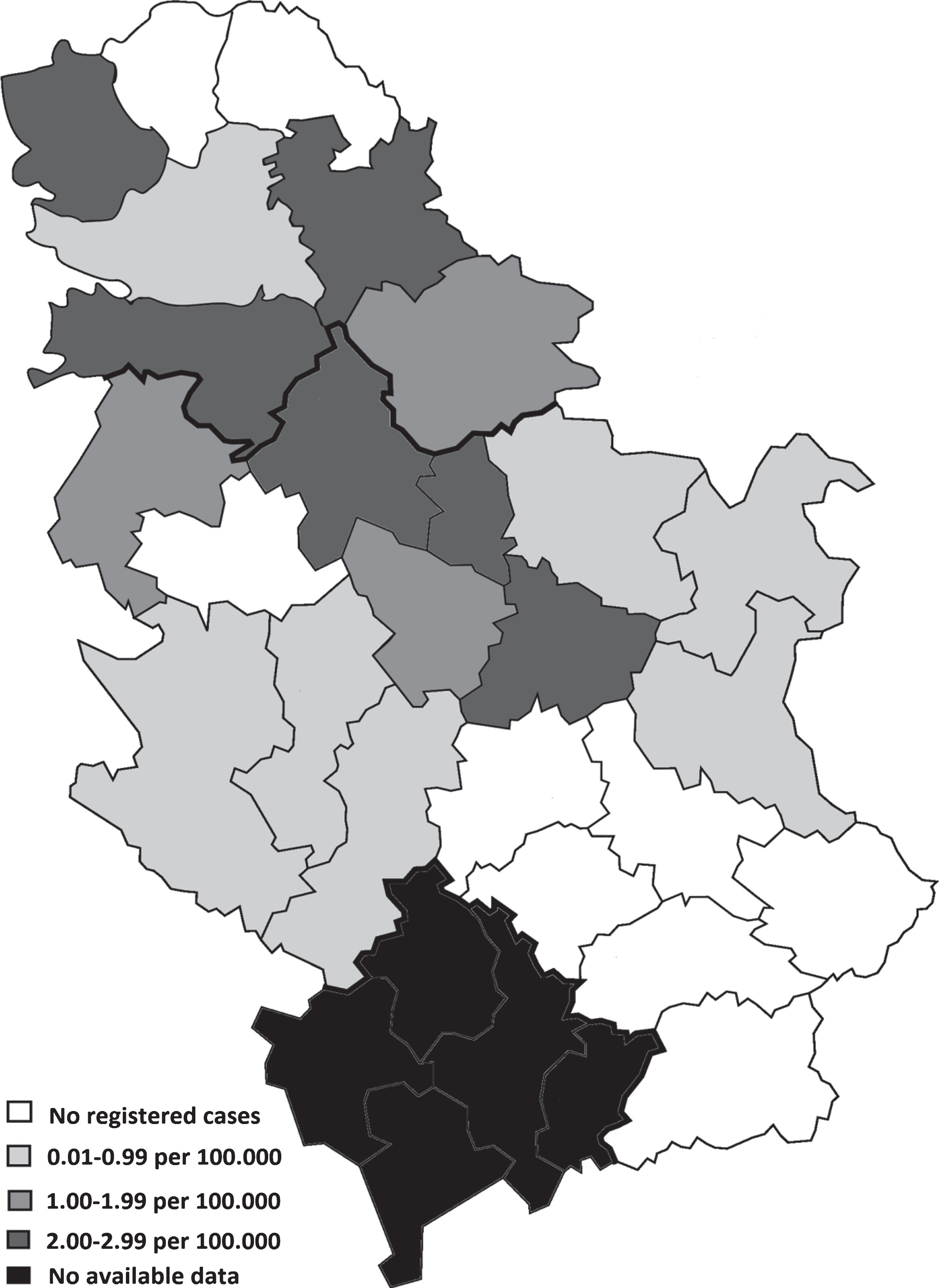
Number of registered patients in the regions of Serbia (n = 84).
Sociodemographic, clinical and laboratory characteristics of DM2 patients
The main sociodemographic features are shown in Table 1. In our cohort of DM2 patients, 63.2% (n = 55) were females. DM2 started in the fourth decade of life in majority of patients. The most common first symptom in our patients was lower limb weakness, followed by handgrip myotonia and pain in upper and lower limbs (Table 1). However, 17% of our patients were diagnosed after DM2 diagnosis in a family member although they did not complain of any symptoms originally. In seven (8%) patients of our cohort cataracts preceded muscular symptoms. Extrapyramidal symptoms and signs were the first manifestation of DM2 in three (3.4%) patients (one patient had camptocormia, postural tremor and bradykinesia; another had postural tremor and bradykinesia; and the third one had postural tremor only). All of these patients were examined by a movement disorder specialist and none of them fulfilled criteria for Parkinson disease or some of atypical parkinsonisms. Furthermore, all of them were L-dopa unresponsive. The diagnostic delay was 11.8±11.3 years and mean age at entry in the Registry was 51.0±13.4 years.
Sociodemographic and clinical features of investigated DM2 patients (n = 87)
DM2 - myotonic dystrophy type 2; DM1 - Myotonic dystrophy type 1.
Muscle symptoms and signs in DM2 patients (n = 87)
MRC – Medical Research Council, EMG – electromyography.
Regarding muscle symptoms, cranial and neck muscle involvement was rare and mild, while the most prominent symptoms were observed in proximal muscles of lower limbs (Table 2). In 75.3% of DM2 patients lens opacities were present at time at entry in the Registry, and 42.6% of patients already had cataract surgery (Table 3). Mean age of cataract surgery was around 50 years. Although approximately 20% of patients had bradycardia, severe ECG abnormalities were noted only in 8.3% of DM2 subjects, and EF < 55% was observed in 4.1%. Cardiac pacemaker was implanted in four (4.6%) patients due to: 1. first degree AV block and left bundle branch block, 2. third degree AV block and left bundle branch block after myocardial infarction, 3. Mobitz II, and 4. left bundle branch block. History of angina pectoris was reported in 3.4%, the acute myocardial infarction in 2.3% and bypass surgery in 3.4% of patients. Only 10% of our cohort had pulmonary restriction with FVC < 90%. No one of them had any respiratory symptoms or requested pulmonary therapy, although we did not specifically assessed for sleep breathing disorders. Insulin resistance and diabetes mellitus were frequent in our DM2 cohort (20.7% and 17.2%, respectively).
Clinical and laboratory data of DM2 patients (n = 87)
ECG – electrocardiography, EF – ejection fraction, FVC – forced vital capacity, HDL – high density lipoprotein, LDL – low density lipoprotein.
Gender differences
The average age at onset of DM2 was similar in men and women (33.5±12.1 vs. 39.9±12.0, p > 0.05, respectively) (Table 4). Male DM2 subjects more frequently had: snoring, frontal baldness, sterility and polyneuropathy (p < 0.05). Serum HDL level was lower and glycaemia higher in male DM2 subjects (p < 0.05). Waddling gait and increased reflexes were more common in females (p < 0.05).
Gender differences in patients with DM2 (n = 87)
HDL – high density lipoprotein, *p < 0.05.
Correlations with disease duration
With longer disease duration, we observed progression of weakness in all skeletal muscle groups (p < 0.05). Also, duration of percussion myotonia was longer with longer disease duration (rho = +0.26, p < 0.05). Mean disease duration in patients with severe cardiac abnormalities was 17.5±13.1 years vs. 10.5±11.8 years in patients without cardiac abnormalities (p < 0.05). Moreover, presence of following features is also shown to correlate with disease duration in our study: cataract, polyneuropathy, baldness, constipation and gallstones (p < 0.05).
DISCUSSION
Our study included 87 patients with genetically confirmed DM2. We were probably not able to detect all persons with DM2 gene mutation in Serbia since it is a multisystem disorder which is often hidden in a wide range of mild symptoms and signs. We believe that registered patients represent only the top of the iceberg and they are the most severe cases who required medical treatment. Although there is not a precise prevalence of DM2 in a general population, it is suspected that this disease is more frequent than previously considered. Vanacore et al. noted that the age-standardized prevalence of DM2 in province of Rome (Italy) was almost 1 per 100 000 inhabitants [12], which is very similar to the prevalence of registered DM2 patients in the Republic of Serbia. However, Suominen et al. reported that the prevalence of DM2 genetic mutation is significantly higher (55 per 100 000 inhabitants) in the general population of Finland [13]. It seems that the prevalence of registered DM2 patients in our country is higher than in most other countries, probably due to the,,founder effect” of the mutation and its historical trail. Main goals of the registries should be the increased number of diagnosed cases and reduction of the diagnostic delay which was 11.8±11.3 years in our cohort, similarly to previous data [12]. This could be due to the mild and heterogeneous phenotype of DM2 who are sometimes evaluated by specialists other than neurologists. Moreover, the genetic analysis is still not equally used across our country.
The most common initial manifestations in our cohort were proximal muscular weakness, hand myotonia and myalgia in extremities, which is in accordance with findings of previous studies [14–16]. The rest of our DM2 cohort had a presenting symptom in the form of cataract (8%), which is in accordance with previous data of 5% to 14% of patients who initially had lens opacities [18, 19]. Furthermore, we observed 11 patients with DM2 among 150 patients with early onset cataract and without any neurological symptoms [19]. Extrapyramidal symptoms and signs were noted as an initial symptom in 3.4% of our DM2 patients. In line with this, we have previously found impaired glucose metabolism in thalami and striatum in 38% of DM2 patients using fluorodeoxyglucose positron emission tomography study [20]. Sansone et al. reported a case of a patient with DM2, parkinsonism, and impaired glucose metabolism in parietal and temporal regions, as well as in thalami [21]. Further on, substantia nigra hyperechogenicity was previously observed by transcranial sonography in 20% of our DM2 patients [22]. These data suggest that a certain number of DM2 patients should be recognized by general practitioners, ophthalmologists, specialists of movement disorders and others.
In present study, 8% of our patients had severe ECG involvement, while our earlier research in DM1 (n = 111) indicated that 28.8% of patients had severe ECG abnormalities [23]. In addition, previous studies underline that around 24% of DM1 and 17% of DM2 patients have severe ECG abnormality [24]. Cardiac abnormalities in DM2 appeared to be similar but less frequent than in DM1 [15]. Severe ECG abnormalities were associated with higher risk of sudden death in DM1, and this can also be a case in DM2 [9].
Cataract was present in 75.3% of our patients with DM2, which is slightly above the range of 49–60% of patients reported in previous series [16, 18]. Peripheral neuropathy was diagnosed in 17.2% of our patients, which is in accordance with 23% to 50% in previous studies. We did not observe association between polyneuropathy and presence of diabetes. Polyneuropathy was usually mild and did not worsen the phenotype [16, 25].
Increased LDL cholesterol (98%), total cholesterol (74%) and creatine kinase (49%) levels were the most common laboratory abnormalities in our cohort. It was previously reported that abnormal laboratory values were present in more than half of DM2 patients [26]. All these findings indicate that permanent laboratory monitoring, especially of metabolic impairments, is needed in order to prevent potential complications.
Waddling gait was more common in females with DM2. Similarly, Montagnese et al. showed that proximal muscle weakness was more common in females than in males with DM2 [14]. The explanation could be the fact that men naturally have a higher percentage of muscle mass. The presence of polyneuropathy in our cohort was more frequent in male subjects. We also previously observed more prevalent polyneuropathy in men with DM1 [27]. Sterility was more often in DM2 males compared to females (32% vs. 2%). Occasionally DM2 men present with hypogonadism, but this appears to be more common in DM1 [28]. Our previous results showed that 72% of men with DM1 had erectile dysfunction and hypogonadism [29].
DM2 is a slowly progressive disease, which initially affects the muscular system [14]. In our study weakness of all muscle groups correlated with disease duration. Also, percussion myotonia was more prolonged with the longer disease duration, in contrast to DM1 where myotonia was less pronounced when muscle weakness was profound [17]. It should be noted that variants in chloride and sodium channels are genetic modifiers of myotonia in DM2 [30]. Longer disease duration in our patients also correlated with the occurrence of severe cardiac abnormalities and cataract. Sansone and colleagues reported progression of cardiac involvement during time in DM2 patients, although it was slower and less severe than in DM1 [15]. Thus, permanent monitoring of DM2 patients is needed in order to detect different multisystemic features. It seems that during disease progression, more frequent cardiologic and ophthalmologic check-ups should be performed.
CONCLUSION
Prevalence of DM2 seems to be much higher than previously thought. Initial symptoms are apparent in the fourth decade of life, usually as muscle weakness in proximal lower limbs. However, cataracts and extrapyramidal symptoms may also be the initial symptoms. Severe ECG abnormalities were found in 8% of patients, systolic dysfunction of the left ventricle in 4%, and mild respiratory restriction in 10%. Although rare, cardiorespiratory abnormalities should be carefully screened in order to prevent potentially life threatening complications. Diabetes mellitus was present in even 17% of DM2 cases, and dyslipidemia in virtually all patients. Some gender-specific features were noted in our DM2 cohort. Majority of symptoms showed slow progression during time.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
Footnotes
ACKNOWLEDGMENTS
This study was supported by the Ministry of Education, Science and Technological Development of Serbia (grant #175083).