
Abstract
Background:
The TREAT-NMD Global Registry Network is a global collaboration of neuromuscular disease registries, including myotonic dystrophy type 2 (DM2), which aims to facilitate collaborative research and clinical trials.
Objectives:
This study aimed to assess DM2 patients included in the network, and to analyse their socio-demographic and clinical features.
Methods:
Data were collected through email surveys sent to 16 TREAT-NMD myotonic dystrophy core member registries. 10 registries enrolled DM2 patients.
Results:
The total number of DM2 cases was 1,720, with the Czech, German, and USA registries enrolling the most patients (445, 430, and 339 cases, respectively). The highest rates were seen in Czechia and Serbia (4.2 and 2.0 registered per 100,000 population, respectively). High DM2:DM1 ratios were seen in Central Europe. The median age at registry entry was 51 years. Symptom onset occurred before age 20 in 14% of cases. One fifth of patients used an assistive device to walk, and 4% were non-ambulatory. Insertion of a pacemaker or implantable cardioverter-defibrillator was reported in 4% of subjects, while 7% used non-invasive ventilation.
Conclusions:
This represents the largest DM2 cohort assembled to date, providing demographic and clinical data for future research and trial recruitment, illustrating TREAT-NMD's international reach and the importance of capturing DM2 data.
Introduction
Myotonic dystrophy type 2 (DM2) is a rare, late-onset, multisystem, inherited disorder with a slowly progressive course. It was first described clinically by Ricker et al., 1 and Thornton, Griggs and Moxley 2 in 1994 and later also by Meola et al., 3 and its genetic basis, an unstable CCTG repeat expansion in intron one of the CNBP (cellular nucleic acid-binding protein) gene, was only confirmed in 2001. 4 The main clinical characteristics of the disease are slowly progressive proximal muscle weakness, myotonia, and myalgia. Myotonia is usually less pronounced than in myotonic dystrophy type 1, but severe myotonia could also occur in DM2 because of the modifier genes, i.e., chloride or sodium channels.5,6 Multisystem features of DM2 include ocular, cardiac, pulmonary, smooth muscle, endocrine, and brain involvement.7,8 Prevalence estimates range from 1/100,000 in Italy to 9/100,000 in Germany.9,10 A study in Finland found two DM2 expansions in 4502 anonymous blood donors suggesting that there are many undiagnosed or possibly asymptomatic people as this would give a much higher rate of 44/100,000 (95% confidence interval = 8 to 178/100,000). 11 Historically DM1 has been more widely recognised and thought to be more common, but in the above Finnish study just as many DM2 as DM1 expansions were detected in the same sample. 11
The rarity of DM2, and its wide phenotype spectrum, including several patients with mild disease severity, 12 all mean that our knowledge of the disease needs continuously to be systematically updated. This significantly impacts disease management, including delayed diagnosis, suboptimal patient care, limited access to treatment, and lack of clinical trials for molecular therapies. At the end of the twentieth century, disease-specific patient registries at regional or, preferably, national levels were recognised as essential tools to fill this gap in our knowledge regarding rare diseases. Registries also provide practical structures for collaboration between healthcare providers, researchers, patients, public health authorities, and industry.13–15 The International Rare Diseases Research Consortium (IRDiRC) has recognised this important role of registries and has emphasised the need for them to collaborate in collecting internationally agreed standardised datasets. 16 In keeping with this, a minimum data set for myotonic dystrophy in the adult population was agreed at the European Neuromuscular Centre (ENMC) workshop in Naarden in 2008, sponsored by TREAT-NMD and the Marigold Foundation (the “Naarden dataset”). 17 Collecting this minimum dataset was adopted as a prerequisite for being a myotonic dystrophy type 1 (DM1) core member registry in the TREAT-NMD Global Registries Network. Although this was primarily established for patients with DM1, many registry members adopted the same dataset for collecting data on patients with DM2.
This study aimed to explore the available patient data on DM2 included in the TREAT-NMD Global Registry Network, paying particular attention to their demographic and clinical features and comparing data between different countries.
Materials and methods
A questionnaire in the form of an Excel spreadsheet was sent to the sixteen core members of the TREAT-NMD Myotonic Dystrophy Global Registry Network (Figure 1). The survey included all mandatory and most of the highly encouraged items of the “Naarden dataset” (see above) (Table 1).17,18 The last four highly encouraged items (family history, ethnic origin, participation in another registry, and genetic testing details (name of laboratory, date of the test, method of test, repeat length)) were not included in the survey. Where results were incomplete, follow-up emails were sent to registry curators. Data were acquired over eight weeks in the first half of 2022. In accordance with the Declaration of Helsinki, all patients signed informed consent to be enrolled in the Registries and to share their anonymous data for scientific purposes. This process was approved by the local ethical committees. For this study, only summary report from each registry was used and no patients’ individual data were available, thus there were no possibilities to identify individual patients in any way.
TREAT-NMD registries yas of 2022. World map with TREAT-NMD core registries presented in black.
Core dataset for myotonic dystrophy registries.

ECG – electrocardiography.
FVC – forced vital capacity.
Descriptive statistics were used to analyse the data. Population recruitment rates were calculated using population data from the last census of each country available online at the site of the Statistical Offices.
Results
Out of 16 TREAT-NMD myotonic dystrophy core registries, 13 responded. Three of the responders did not include DM2 patients (Slovenia (an essential paediatric registry), Japan (no patients having been diagnosed), and Turkey). We present here the data from the ten registries that do collect data from DM2 patients.
The present study covered a mixed population of more than 500 million from Europe, Oceania, and North America and provided data on 1720 DM2 patients. Thus, this is the largest cohort of DM2 patients reported to date. The number of registered DM2 cases compared with DM1 cases is shown in Table 2. Although DM2 was 2.5 times less frequently reported than DM1 overall, Central European countries reported similar or even higher numbers of DM2 cases. The DM2 diagnosis was genetically confirmed in 81% of the patients. Genetic diagnosis rates varied among registries. All patients without confirmed genetic diagnosis had clinical presentation of DM2 and at least one first-degree relative or parent with DM2. In patient-reported registries in some cases genetic diagnosis was not entered in the Registry by patients themselves, although they were genetically tested and had confirmed DM2.
Registered cases of myotonic dystrophy in the core registries of the TREAT-NMD Global Registry Network.
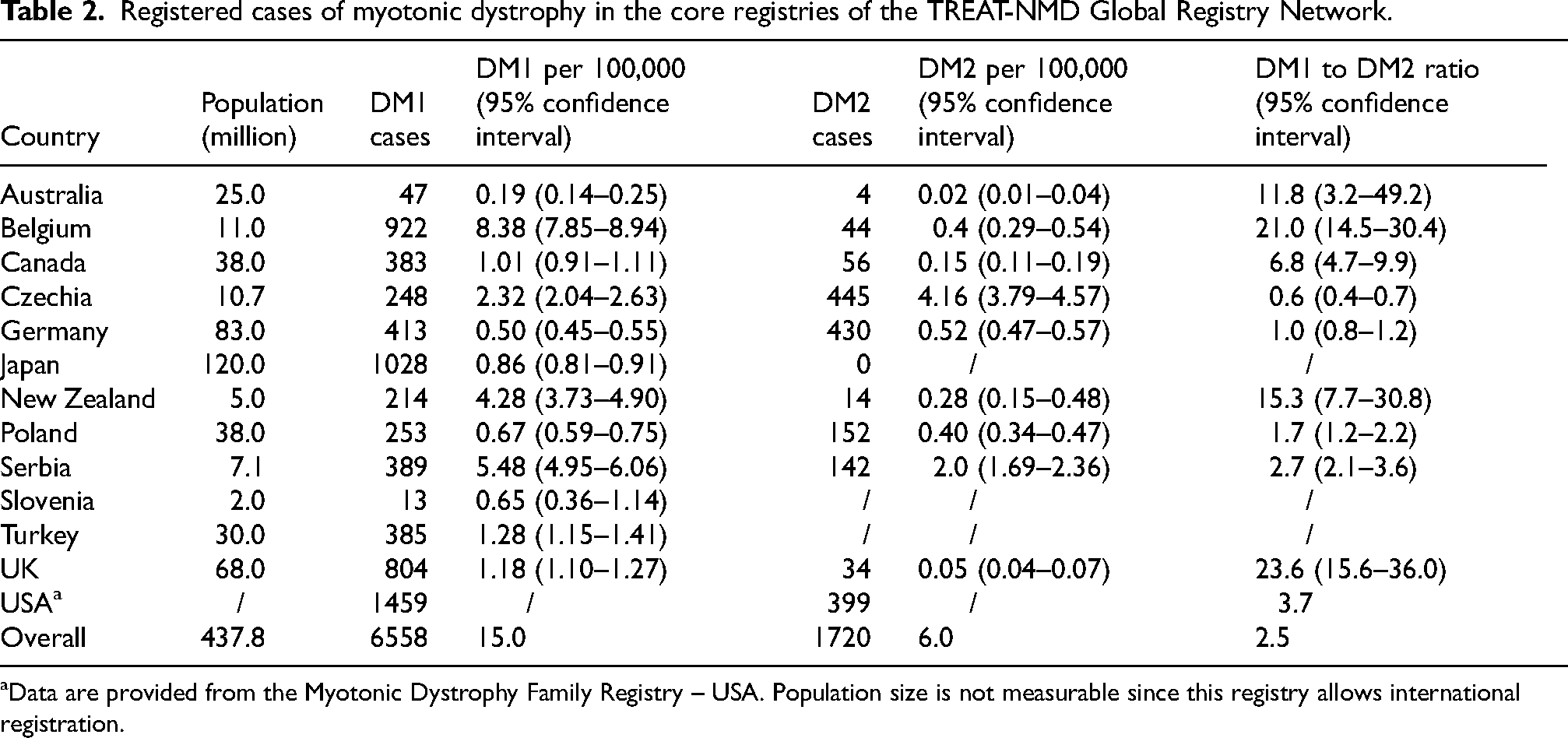
Data are provided from the Myotonic Dystrophy Family Registry – USA. Population size is not measurable since this registry allows international registration.
Female patients comprised 63% of the sample, with female predominance in all ten registries. Symptom onset occurred in adulthood in 83% - with about half of these in the 21 to 40 age group; 14% had juvenile (<20 years) onset and a further 3% were reported as genetically proven gene expansion carriers but were asymptomatic (Table 3). The median of mean age (per registry) at entering a registry was 51 years, and at the time of reporting, the median age was 56.5 years.
Symptom onset in patients with DM2.
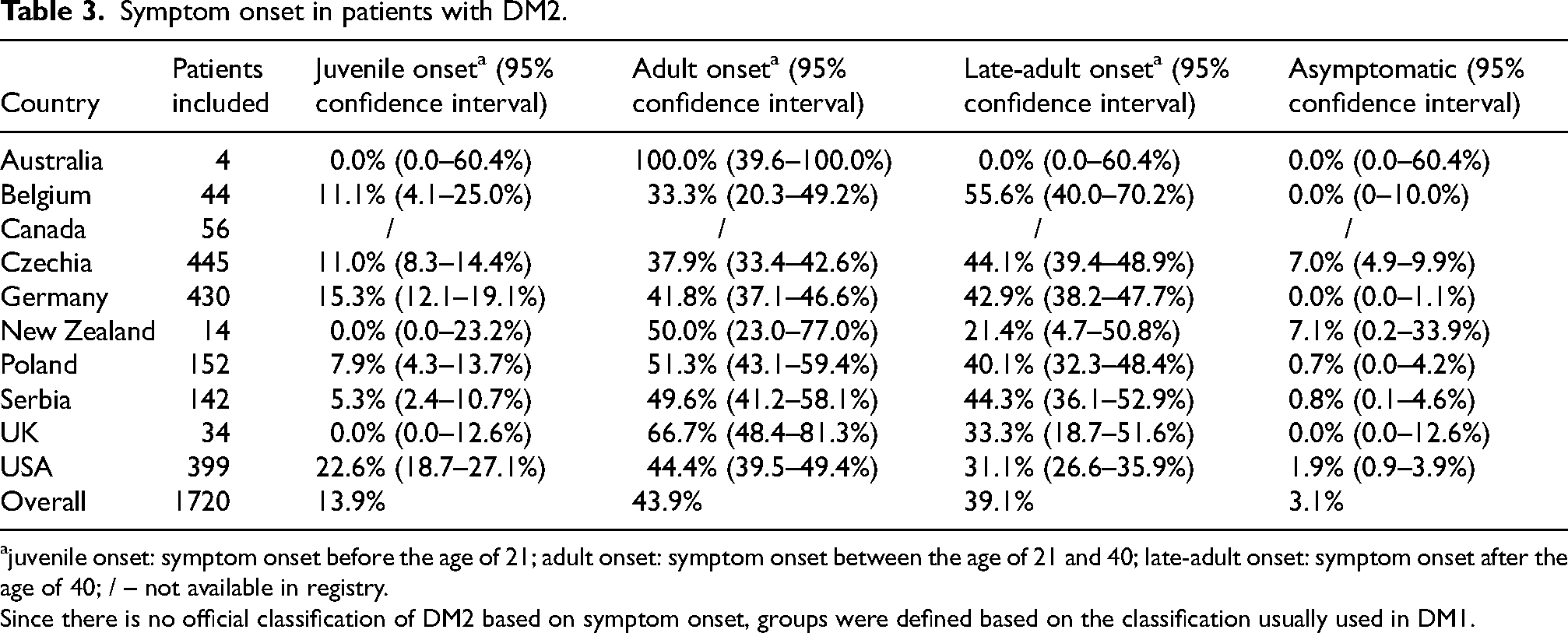
juvenile onset: symptom onset before the age of 21; adult onset: symptom onset between the age of 21 and 40; late-adult onset: symptom onset after the age of 40; / – not available in registry.
Since there is no official classification of DM2 based on symptom onset, groups were defined based on the classification usually used in DM1.
The use of a walking aid was reported in 20% of patients, while 4% were not ambulatory. Noticeable differences regarding ambulation status among registries were observed: 95% of the Serbian patients were reported as walking unassisted compared with just half of the patients in the American registry (Figure 2). Hand grip myotonia was a core phenotypic feature in at least half of the patients in each registry and in 70% of DM2 patients overall.
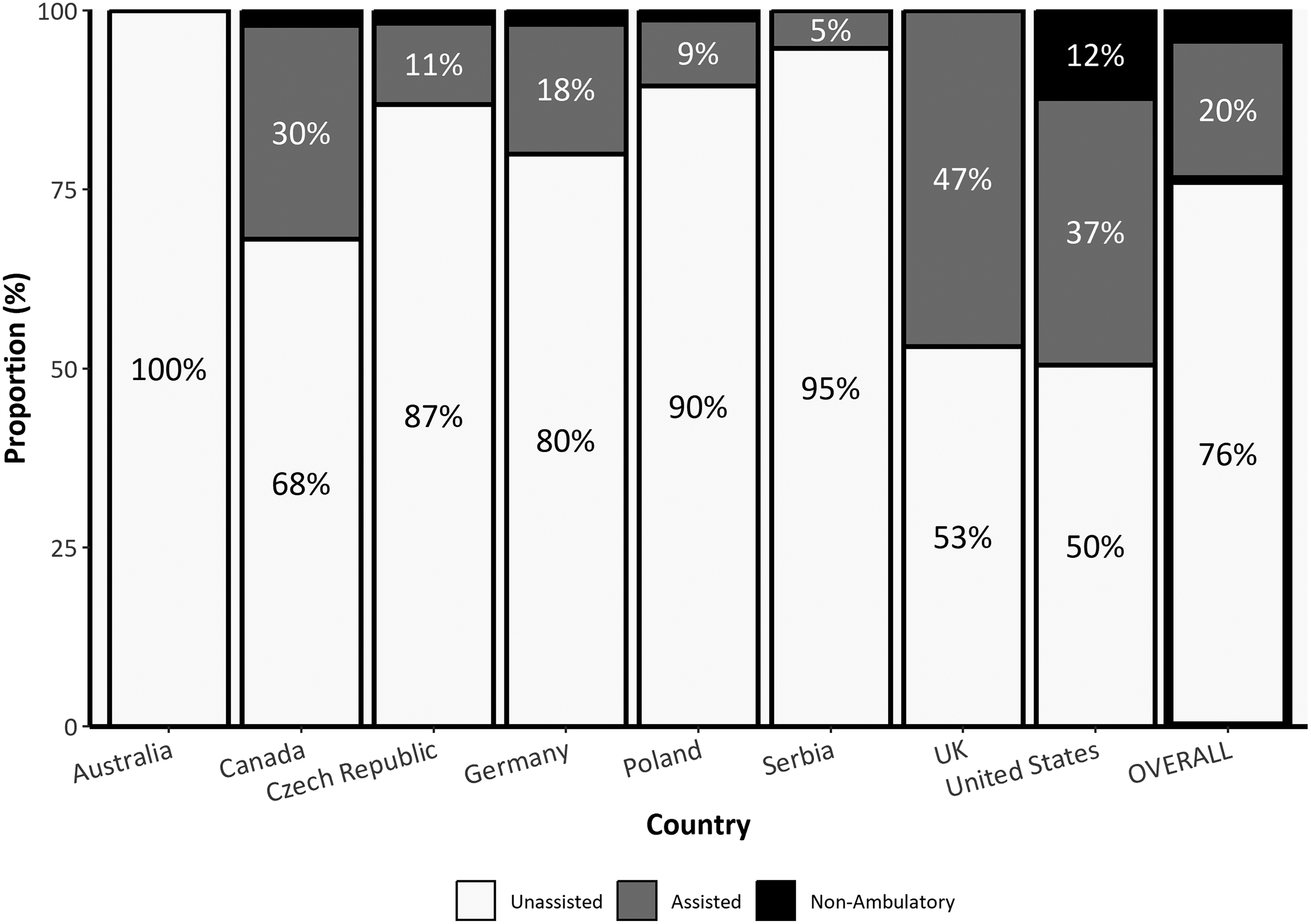
Ambulation status of patients with DM2.
Cataracts/cataract surgery was reported in 58% of the cases overall and was the most common reported extramuscular manifestation of the disease. However, its frequency differed widely between countries, reported in just one-third of German patients, in contrast to 73% of patients in Serbia. One out of four patients (26%) had different types of cardiac conduction disturbances or arrhythmias, leading to pacemaker or cardioverter-defibrillator (ICD) implantation in 4% of cases. Reported rates of cardiac involvement and implanted cardiac devices also varied widely (Figure 3). A restrictive pattern in pulmonary function studies defined as forced vital capacity (FVC) below 90% was another disease-related feature, present in one-third (34%) of the patients. Respiratory support was being used by 7.4% of patients (non-invasive ventilation – NIV in 6.7% and invasive ventilation in 0.7%). Spirometry findings and use of mechanical ventilation again varied markedly between countries, with none of the Serbian and Polish DM2 patients receiving support, through to 17 to 20% of the cases in American and UK registries (Figure 4). Daytime sleepiness was reported in 40% of DM2 patients. One in five subjects (22%) complained of swallowing difficulties, although a nasogastric tube was occasional (0.5%). Significant differences in reporting rates were also observed regarding this symptom, with dysphagia reported in 7.1% of the Czech patients and as high as 37.5% in the UK data.
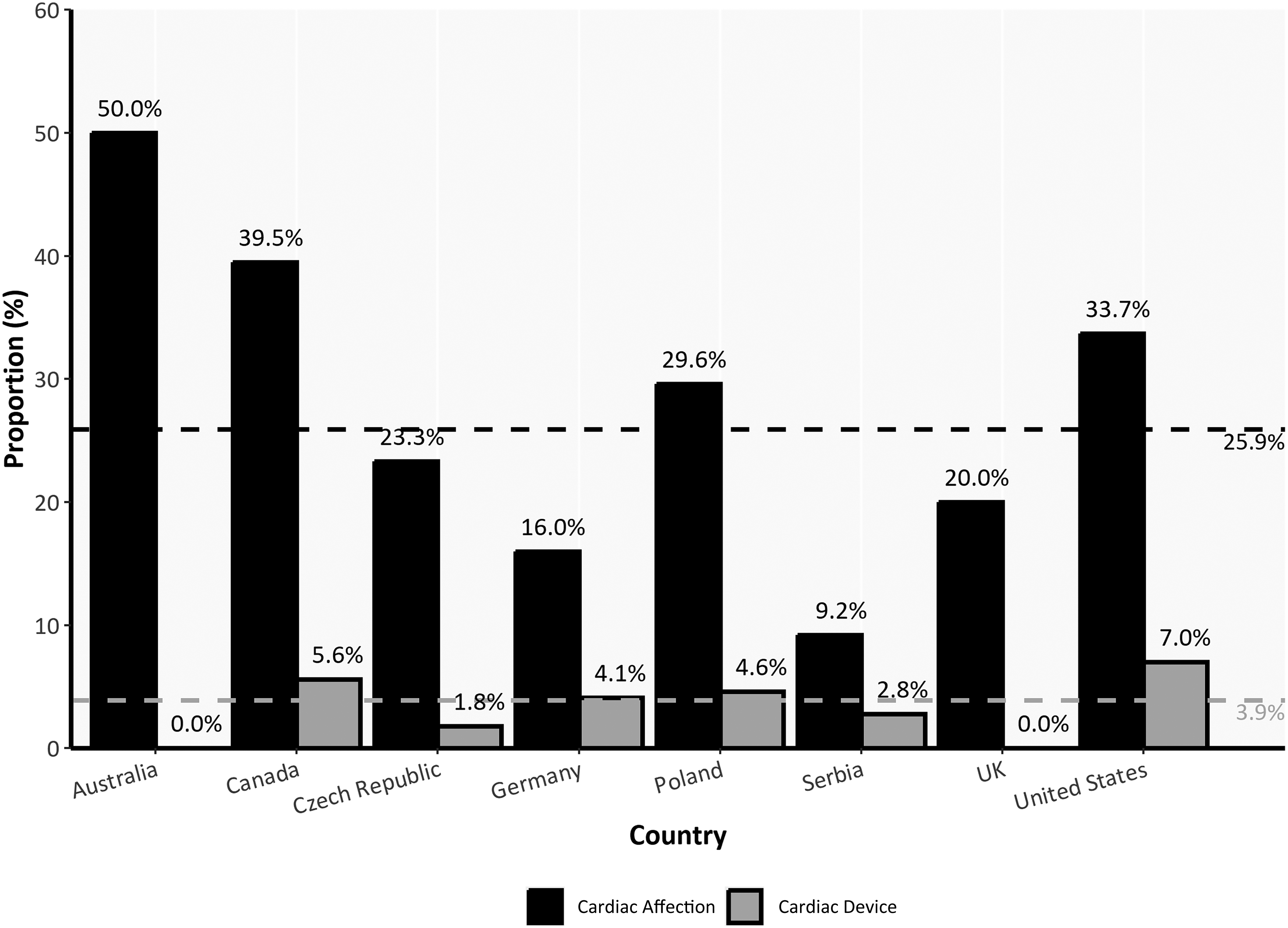
Percentage of DM2 patients with cardiac conduction defects/arrhythmias and cardiac device. Cardiac involvement – presence of cardiac conduction defects / arrhythmias; Cardiac device – presence of cardiac pacemaker or ICD (implantable cardioverter-defibrillator); Dashed lines indicate value of two variables for the overall DM2 cohort (black for cardiac affection, grey for cardiac device).
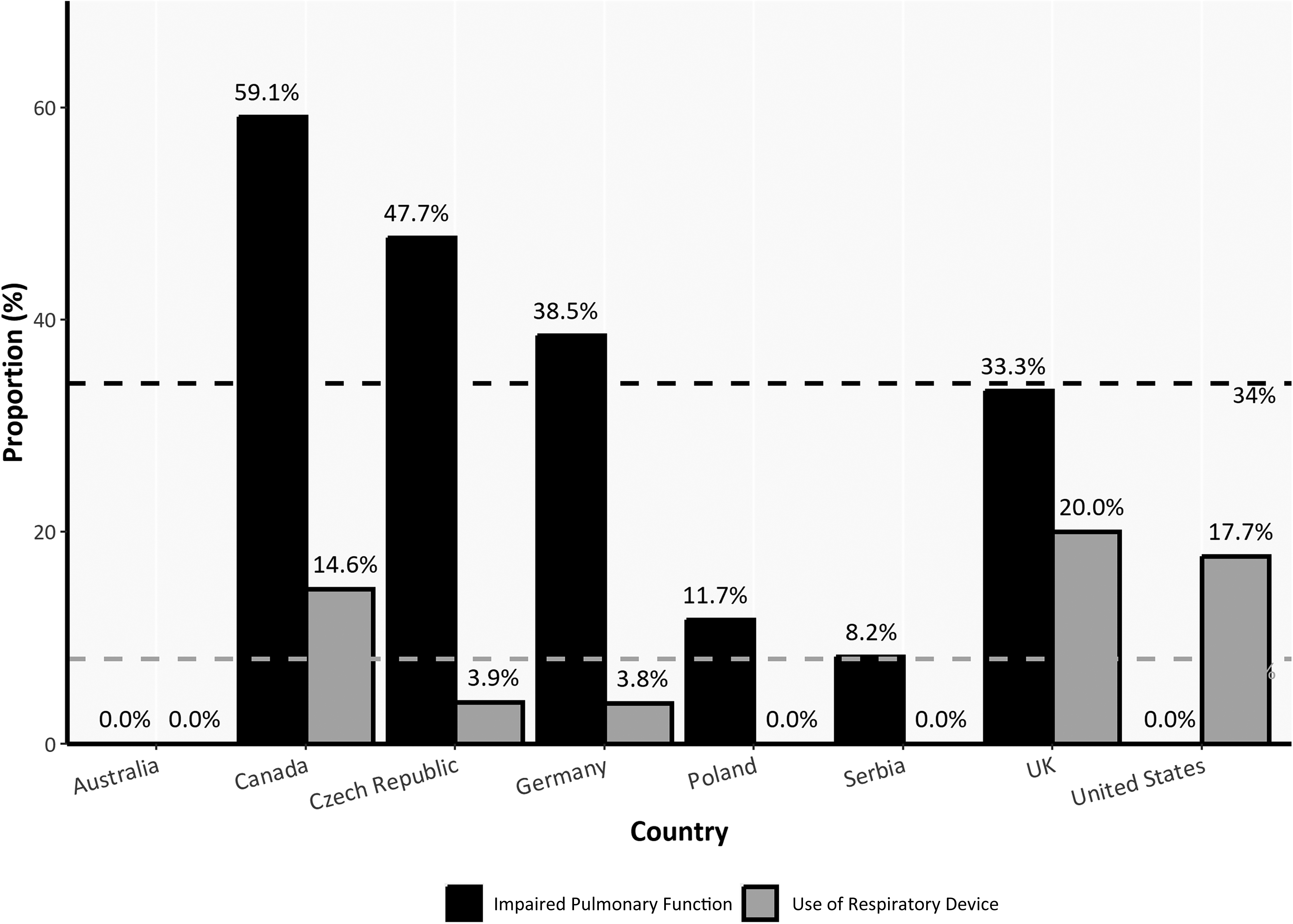
Percentage of DM2 patients with impaired pulmonary function and those who use respiratory support devices. FVC – forced vital capacity; Respiratory support – NIV (non-invasive ventilation) or IV (invasive ventilation); dashed lines indicate median value of two variables for all registries (black for FVC, grey for respiratory support).
Data were available from the Czech and Serbian registries on twelve patients who had died after entering the registry. The following causes of death in DM2 were identified: malignancy in six patients (n = 6), stroke (n = 2), brain haemorrhage (n = 1), bronchopneumonia (n = 1), unspecified lung infection (n = 1), and ileus (n = 1).
Discussion
An increasing number of global collaborative initiatives in rare diseases have highlighted the importance of rare disease registries as the important promoter in developing effective treatments for these conditions. Key principles have been proposed for their establishment, including agreeing on minimum datasets.16,18,19 Based on an agreed and harmonised dataset for DM1,17,18 we present the first multinational collaboration for DM2.
This report provides the sociodemographic and clinical data of more than 1700 patients living with DM2. This is the largest multinational DM2 cohort ever reported. Although registries are not designed to provide accurate disease prevalence, our dataset does provide a minimal prevalence as high as 4.2/100,000 in Czechia and 2.0/100,000 in Serbia. These results are consistent with previous reports from Italy and Germany, though lower than the rate found in Finland (55/100,000).9–11 Rates were lower in Western Europe, for example, just 0.4/100,000 in Belgium, with lower rates in the UK, Australia, Canada, and New Zealand. Initially, this might be considered to be due to poorer case ascertainment. Still, the DM1:DM2 ratios, especially the consistent cline in this ratio across Europe, and previous similar reports10,11 suggest that these numbers reflect genuine differences - potentially due to a DM2 expansion founder effect from the DM2 expansion in central Europe. 20 Indeed, former haplotype and linkage disequilibrium studies have suggested that the DM2 expansion originated from a single or a few founder mutations.4,21,22 In one study, the DM2 expansion mutation was estimated to have arisen 200–540 generations ago. 22 In another study, a family of apparent Afghan/Tajik ancestry was shown to share the common European haplotype, suggesting that the DM2 expansion occurred before the migration of Indo-Europeans into ancient Afghanistan in 2000–1000 BC.4,23
Four out of five (81%) patients in the registries received genetic confirmation of the disease, with a difference among countries. All patients without confirmed genetic diagnosis had clinical presentation of DM2 and at least one first-degree relative or parent with DM2. In patient-reported registries in some cases genetic diagnosis was not entered in the Registry by patients themselves, although they were genetically tested and had confirmed DM2. This makes percentage of genetically confirmed patients even higher. This number has increased compared to the previously published data. 24 We believe that in the era of increasing disease awareness and high availability of genetics testing, molecular diagnosis would facilitate the recruitment of patients into clinical research, particularly for ongoing and future therapeutic studies.
Our study showed female predominance in all DM2 registries included in the study, as previously reported by others.9,25,26 Female hormones may affect how the disease manifests, as it has been reported that pregnancy may worsen the disease and even be a trigger for the first symptoms to appear.27,28
Most of our patients had adult -onset of the disease, which is also consistent with the literature.10,24–26,29 However, whereas juvenile-onset was previously considered very rare, our sample shows that 14% of patients had symptom onset before age 20. Further research is needed to these juvenile cases of DM2. A low prevalence of asymptomatic individuals with the CNBP expansion (3%) was noted. Suppose these patients presented for a thorough clinical neuromuscular exam supported by investigations such as measurement of creatine kinase levels, gamma-GT, echocardiogram, and slit lamp assessments, in that case, many of them might have clinically detectable disease signs and laboratory abnormalities although still without any complaints.30–32
Up to a quarter of DM2 patients from our registries were non-ambulatory (4%) or required support to walk (20%) though the rates varied widely between countries. The lowest number of patients with independent gait was noted in those populations where registry data is entirely (USA) or partially (UK) patient-reported. The availability of walking aids in different countries may also contribute to these discrepancies. 33 Hand grip myotonia was present in nearly 70% of our patients, which aligns with rates of 70–90% reported in previous case series.34,35
More than half of the patients had cataracts making this the most common extramuscular disease manifestation in our cohort. However, substantial international differences were again noted. As a recognised hallmark of DM2, some authors propose that DM1 and DM2 genetic screening should be undertaken in patients with early-onset cataracts of undetermined aetiology, even when they lack any muscle-related symptoms. 32 In the Serbian population, 7% of such patients had expansions in the CNBP gene. 32 The presence of cataracts was also the most important item of the recently proposed early diagnostic score for DM2 (DM2-EDS). 36
Different types of cardiac conduction defects and/or arrhythmias were found in more than a quarter of DM2 patients in the present study, and the most severe cases had received a cardiac pacemaker or ICD, consistent with previous reports.25,37,38 Higher frequencies of conduction abnormalities and cardiac arrhythmias compared with the general population, in addition to occasional cases of sudden cardiac death 23 or severe cardiomyopathy requiring cardiac transplant, 39 have all been linked to the CNBP expansion. This clearly indicates the importance of detailed cardiological follow-up for DM2 patients. In some registries higher percentage of cardiac impairments with relatively lower percentage of implanted devices were reported. This may be related to the fact that in some countries mild cardiac conduction defects were entered in the registry as pathological. Also, in certain countries more extensive cardiac evaluation is probably done. Finally, different countries have variable thresholds to place devices in response to cardiac conduction defects and/or arrythmias. This suggests that international consensus should be made for detecting cardiac impairments and for placing devices in DM2.
Mild respiratory involvement was also a frequent finding in our cohort, although, in keeping with previously reported rates of 6–15%, 38 only 7% of patients used mechanical ventilation. 40 A significant drop (<20%) of FVC and FEV1 between sitting and supine position have been recognised as a predictor of poor respiratory outcome and linked with ventilatory restriction, hypoxaemia, and hypercapnia in patients with DM2. This has led to the recommendation that DM2 patients should have regular pulmonary check-ups to assess pulmonary function in both sitting and supine positions.40,41 Further, semi-structured interviews with open-ended questions have revealed subjective breathing difficulties in half of the DM2 patients, 42 indicating the relevance of obtaining patient-reported assessments of respiratory involvement to supplement objective pulmonary assessments. In our study, there were pronounced differences in the reported rates of mechanical ventilation, with none of the patients in the Serbian and Polish reporting receiving this, whereas, at the upper end, 20% of UK patients used it. We believe that the difference should be a call to action for experts to provide international disease-specific therapeutic recommendations for this patient group.
Dysphagia was a recurrent feature in our cohort, present in more than 20% of the cases, although with just the occasional nasogastric tube placement (0.5%). Again, there was variation in the reported rates of dysphagia among the different registries, and overall, this was slightly lower compared with previously reported rates of 34 to 52%.43–45 Dysphagia and other gastrointestinal symptoms (constipation, abdominal pain) require high awareness and, where present, may indicate the need for a specific gastrointestinal questionnaire and the specialist to be included in the patient's management.
TREAT-NMD member registries assembled the largest international DM2 cohort, providing important clinical and demographic data. Although most patients had adult late-onset disease, clinicians should be aware that DM2 may occur before the age of 20. Since age at symptom onset was reported by patients themselves, this may be a significant limitation. Sometimes patients might not know what symptoms are associated with DM2 or they do not properly recall when symptoms started since DM2 is a slowly progressive disorder.
DM2 is more common in Central/Eastern Europe. Up to one quarter of the patients walked supported or were not ambulatory at all. Cardiac, pulmonary, and gastrointestinal involvement was recurrent, indicating the need for a multidisciplinary patient approach and management.
This study is not without limitations. Registry patients are not an ideal representation of the DM2 population. Also, only select countries have a registry. For instance, no South American countries were represented. The conclusions from this paper are limited by these facts.
International myotonic dystrophy registries should aim to capture both DM1 and DM2 data, to contribute to this important collaboration. This will improve our disease knowledge, allowing an assessment of the global disease burden and crucially act as a tool to support future research and clinical trial recruitment.
Footnotes
Acknowledgments
The authors have no acknowledgments to report.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
Raw data were generated at each registry included in this manuscript. Derived data supporting the findings of this study are available from the corresponding author on request.