
Abstract
Background:
Cerebrovascular dysfunction confers risk for functional decline in Alzheimer’s disease (AD), yet the clinical interplay of these two pathogenic processes is not well understood.
Objective:
We utilized Alzheimer’s Disease Neuroimaging Initiative (ADNI) data to examine associations between peripherally derived soluble cell adhesion molecules (CAMs) and clinical diagnostic indicators of AD.
Methods:
Using generalized linear regression models, we examined cross-sectional relationships of soluble plasma vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), and E-Selectin to baseline diagnosis and functional impairment (clinical dementia rating sum-of-boxes, CDR-SB) in the ADNI cohort (n = 112 AD, n = 396 mild cognitive impairment (MCI), n = 58 cognitively normal). We further analyzed associations of these biomarkers with brain-based AD biomarkers in a subset with available cerebrospinal fluid (CSF) data (n = 351). p-values derived from main effects and interaction terms from the linear regressions were used to assess the relationship between independent and dependent variables for significance (significance level was set at 0.05 a priori for all analysis).
Results:
Higher mean VCAM-1 (p = 0.0026) and ICAM-1 (p = 0.0189) levels were found in AD versus MCI groups; however, not in MCI versus cognitively normal groups. Only VCAM-1 was linked with CDR-SB scores (p = 0.0157), and APOE ɛ4 genotype modified this effect. We observed independent, additive associations when VCAM-1 and CSF amyloid-β (Aβ42), total tau, phosphorylated tau (P-tau), or P-tau/Aβ42 (all < p = 0.01) were combined in a CDR-SB model; ICAM-1 showed a similar pattern, but to a lesser extent.
Conclusion:
Our findings indicate independent associations of plasma-based vascular biomarkers and CSF biomarkers with AD-related clinical impairment.
Keywords
INTRODUCTION
Although amyloid deposition is considered to be the central inciting event in the development of clinical Alzheimer’s disease (AD) [1], age-associated cerebral vascular disease and AD have long been recognized as frequently co-morbid entities. Abundant contemporary epidemiologic data point toward clinically relevant interrelationships between vascular dysfunction and AD, as evidenced by a strong associations between mid-life vascular risk factors and later-life clinical AD [2–9]. Mid-life vascular risk factors have moreover been linked to later-life amyloid burden [10, 11], including in those with high polygenic risk for AD [10]. In support of this narrative, declining dementia incidence in developed countries is thought to be related to general improvements in preventive disease management [12].
Despite these findings and consistent neuropathological observations of highly prevalent mixed pathology in the elderly [13], the question of whether concurrent cerebral vascular disease and AD pathologies represent biologically intertwined or parallel but non-synergistic processes is difficult to fully elucidate and has been a topic of frequent historical debate [13–24]. Given the critically unmet need for effective preventive and therapeutic strategies for AD and the modifiable nature of vascular risk factors, disentangling various pathogenic processes that underlie risk for age-related cognitive decline and identification of biomarkers that may be sensitive to modifiable vascular-mediated risk is of high clinical import.
The majority of prior studies analyzing contributions of co-existing cerebrovascular and AD pathology have found independent and/or additive rather than interactive associations to risk for cognitive decline; however, most studies to-date have largely focused on relationships of imaging-based white matter disease burden to amyloid pathology [23, 26]. In contrast, results from more recent studies using composite [27] or expanded [28, 29] cerebrovascular biomarker panels or AD biomarkers that are inclusive of amyloid, tau, and neurodegenerative pathologies [26, 30] suggest that some synergistic aspects of vascular-mediated associations to AD pathogenesis may have been previously underappreciated.
We sought to utilize peripherally derived molecular measures of vascular endothelial dysfunction as a means by which to assess for potentially more nuanced contributions of vascular risk to AD-asso-ciated pathologies and clinical AD. Cell adhesion molecules (CAMs) are expressed on the vascular endothelial surface and facilitate blood-borne leukocyte recruitment and trafficking across the vas-cular endothelium to sites of tissue damage via stereotyped stages: initial leukocyte tethering and rolling along the endothelial vessel wall (L-Selectin, P-Selectin, E-Selectin, and vascular cell adhesion molecule-1 (VCAM-1)), followed by firm adhesion (intercellular adhesion molecule-1 (ICAM-1), VCAM-1), and eventual chemotaxis and transendothelial cell migration (PECAM; Platelet And Endothelial Cell Adhesion Molecule) [31–36]. Membrane-bound CAMs are subsequently shed as soluble forms after a time delay, presumably as part of a feedback mechanism [37], and the soluble fraction can be conveniently measured in blood samples to detect processes of active inflammation involving the vascular endothelium.
Elevated circulating and/or cerebrospinal fluid (CSF) CAMs have been reported in relation to a variety of vascular risk factors and cardiovascular disease [38–44], cerebrovascular disease [45], neuroimmunological disorders such as multiple sclerosis [46, 47], and a broad spectrum of immune-mediated disorders including lupus erythematosus, rheumatoid arthritis, asthma, and cancer [39]. There have been a limited number of studies to-date assessing relationships of blood- or CSF-borne CAMs to cognitive aging and dementia in humans [32, 49–56]. However, samples sizes of blood-borne CAMs in most studies have been relatively small, and to our knowledge no studies thus far have tested for interactive associations between soluble plasma CAMs and CSF AD biomarkers in association with the clinical AD spectrum phenotype in a well-characterized AD cohort.
In this study, we analyzed cross-sectional associations of soluble plasma VCAM-1, ICAM-1, and E-Selectin to baseline diagnosis and functional severity staging in Alzheimer’s Disease Neuroimaging Initiative (ADNI) participants. ADNI is a highly selective cohort that has been specifically enriched for biomarker studies of AD-related cognitive decline [63]. Utilizing the three bloodborne CAMs that were available in ADNI plasma proteomics data, we hypothesized that concomitantly higher plasma CAM levels would correlate with severity of baseline diagnosis and functional impairment along the prodromal-to-clinical AD dementia spectrum and tested for modifying effects of Apolipoprotein E (APOE) genotyping on functional impairment. Based upon suggestions of interactive associations of CAMs and AD pathology in recent experimental models, we also hypothesized peripheral CAMs would have a synergistic relationship with brain-based AD biomarkers in influencing baseline functional impairment, which we assessed using CSF biomarkers of AD pathology and neurodegeneration in a subset of subjects for whom those data were available.
METHODS
Participants
Data used in this study were obtained from data of the original ADNI cohort (http://adni.loni.usc.edu). ADNI is a multi-site, longitudinal observational study led by Principal Investigator Michael W. Weiner, MD, that was initiated in 2004 as ADNI-1, and has been extended by successive renewals to the current ADNI-3 cohort that was launched in 2016. The primary goal of ADNI since its inception has been to clarify the roles of imaging and other biomarkers in AD clinical progression in order to validate their use in AD clinical trials [63]. Written informed consent was obtained for all participants, and prior Institutional Review Board approval was obtained at each participating institution. Up-to-date information regarding ADNI can be found at http://www.adni-info.org.
Clinical and cognitive assessment
All subjects underwent an extensive clinical diagnostic evaluation, including basic mental status, neu-ropsychological, physical, and neurological exam-inations. A full description of assessment used can be found at http://adni.loni.usc.edu/methods/documents/
Dementia severity was graded by the Clinical Dementia Rating (CDR) Scale [64, 65], a measure that is widely employed in AD clinical care and research to quantify functional impairment. The CDR is derived through interview with patients and informants and consists of 6 domains (memory, orientation, judgment and problem solving, community affairs, home and hobbies, and personal care), each of which are rated on a 5-point scale (0, no impairment; 0.5, questionable impairment; 1, mild impairment; 2, moderate impairment; and 3, severe impairment). In clinical practice, the algorithm-generated global CDR score produces a total possible score of 0 to 3, denoting a global level of functional status from no impairment (global CDR 0) to severe impairment (global CDR 3) using the descriptors noted above for the individual domain box scores. The CDR sum of boxes score (CDR-SB), by contrast, utilizes a summary of the individual domain box scores and yields a total score of 0 to 18 (higher scores indicating greater impairment), and is frequently used in AD research given greater sensitivity in dementia staging and tracking of progression over time [66].
All participants were given diagnoses of cognitively normal (CN), mild cognitive impairment (MCI), or probable AD; for participants with MCI (global CDR score of 0.5), the inclusion criterion was an amnestic type (CDR memory domain box score of at least 0.5), to specifically ensure enrichment of the cohort with participants at high risk for conversion to AD. All AD patients satisfied NINCDS-ADRDA diagnostic criteria [67] for probable AD and had questionable to very mild dementia (global CDR score of 0.5 but considered borderline dementia) or mild (global CDR score of 1) dementia. Additionally, ADNI was specifically designed to minimize non-AD related risk factors for cognitive impairment or dementia, including vascular dementia. Inclusion criteria for ADNI-1 included a modified Hachinski ischemic score (MHIS) [68] of 4 or less to limit potential contributions from cerebrovascular disease; previously published baseline characteristics for ADNI-1 noted no significant difference between CN, MCI, and AD groups with respect to MHIS [69].
Biomarkers
ADNI proteomics data were collected for a subset of ADNI-1 (the original ADNI cohort) participants who enrolled in this sub-study at baseline. The multiplex panel was based upon Luminex immunoassay technology and had been developed by Rules Based Medicine (MyriadRBM) to measure a range of inflammatory, metabolic, lipid, and other disease relevant indices. A 190-analyte, plasma-based panel of biomarkers previously reported to be related to cell-signaling or disease processes such as AD, meta-bolic disorders, inflammation, cancer, and cardiovascular disease were analyzed through the Biomarkers Consortium Project “Use of Targeted Multiplex Proteomic Strategies to Identify Plasma-Based Biomarkers in Alzheimer’s Disease,” 146 of which met quality control standards. Biomarkers of vascular endothelial dysfunction used in this study included baseline peripheral blood-derived VCAM-1, ICAM-1, and E-Selectin. Further details regarding ADNI proteomics procedures can be found in the data primer, “Biomarkers Consortium Project: Use of Targeted Multiplex Proteomic Strategies to Identify Plasma-Based Biomarkers in Alzheimer’s Disease” (http://adni.loni.usc.edu/wp-content/uploads/2010/12/BC-Plasma-Proteomics-Analysis-Plan.pdf).
A subset of ADNI participants also underwent a lumbar puncture sub-study at the time of peripheral blood collection at baseline. CSF specimens for biomarkers were processed by the Biomarker Core of ADNI at the Translational Research Laboratory, Department of Pathology & Laboratory Medicine at the University of Pennsylvania Medical School, under the direction of Drs. Leslie M. Shaw and John Trojanowski. The Luminex multiplex immunoassay platform was used for measurements of amyloid-β, 42-residue peptide (Aβ42), total tau (T-tau), and phosphorylated tau (P-tau). Over 50 studies have demonstrated clinical sensitivity and specificity for these biomarkers at greater than 80%each. Further details regarding ADNI CSF Biomarker Core procedures can be found in the data primer, “An Overview of the first 8 ADNI CSF Batch Analyses” (http://adni.loni.usc.edu/methods/documents/).
APOE genotyping was performed at screen-ing using established protocols, the details of which can be found at http://adni.loni.usc.edu/methods/documents/.
Statistical analyses
Distributions of peripheral blood levels of CAMs (VCAM-1, ICAM-1, and E-Selectin) and CSF biomarkers (Aβ42, T-tau, and P-tau) for the entire sample were assessed for normality. Using the Shapiro-Wilk test, all plasma CAM and CSF data were found to have non-normal distributions (all p≤0.005). Thus, all plasma and CSF biomarkers were log-transformed for subsequent analysis, and then back transformed for graphical depictions.
A generalized linear model was used to assess VCAM-1, ICAM-1, and E-selectin concentrations (log normal) for each level of baseline diagnosis (CN, MCI, or AD). Using a generalized linear model (the GLIMMIX procedure), CDR-SB and the CDR memory box sub-scores (CDR-Mem) were modeled as binomial distributions by each dependent variable (VCAM-1, ICAM-1, and E-Selectin). We hypothesized that associations between the CDR-SB and each of the CAMs would be greater among those with the highest risk for AD, so we conducted additional analyses to evaluate effect modification by selected known risk factors for AD (e.g., APOE genotype: APOE ɛ4 non-carrier, APOE ɛ4 heterozygote, or APOE ɛ4 homozygote), age, sex, and family history of AD). This would allow us to understand if the relationship between CDR-SB and each of the CAMS was being driven by factors other than our main independent variables. Interaction terms were tested for significance and included in the model when significant.
Using a generalized linear model (the GLIMMIX procedure), CDR-SB was modeled as a binomial distribution by the combination (fit plane) of peripheral plasma CAMs and CSF AD biomarkers (CSF Aβ42, T-tau, P-tau, and P-tau/Aβ42 ratio, all log normal distributions). To understand if effects were interactive or additive, interaction terms were tested for significance and included if appropriate. Individual p-values of the dependent variables in the model were used to test the contribution of these variables. Interaction terms and correlations between CAMs and CSF biomarkers were assessed as a check against concerns over multi-collinearity.
Familywise alpha was maintained at 0.05 using the Holm adjustment for multiple comparisons where appropriate (adjusted p-values are reported, unless otherwise stated). Significance level was set at 0.05 a priori. Classic sandwich estimation was used to adjust for any model misspecification. All statistical analyses were performed using SAS version 9.4 (The SAS Institute; Cary, NC).
RESULTS
Participants
Data were derived from the original ADNI cohort (ADNI-1), which consisted of approximately 200 people with early AD, 400 people with MCI, and 200 CN older individuals. A cohort of 566 ADNI-1 participants (n = 112 AD, n = 396 MCI, n = 58 CN) for whom proteomics data were available were included in the current study. Baseline demographic data including age, sex, and education level (summarized in Table 1), showed expected diagnosis-related characteristics with respect to CDR-SB and CDR-Mem scores. In our subset of the ADNI-1 cohort, we observed the same discrepancy related to higher proportion of male participants in MCI versus CN and AD groups that had previously been reported for the entire ADNI-1 cohort [69]. There were no major differences in vascular risk (as measured by the MHIS) between CN, MCI, and AD groups that would be expected to confound our analyses (Table 1).
Data summary for ADNI-1 participants included in study
AD, Alzheimer’s disease; ADNI, Alzheimer’s Disease Neuroimaging Initiative; CDR-Mem, clinical dementia rating-memory subscale; CDR-SB, clinical dementia rating-sum of boxes; CN, cognitively normal; CSF, cerebrospinal fluid; E4, APOE ɛ4 allele; 1 IQR, first interquartile range; 3 IQR, third interquartile range; MCI, mild cognitive impairment; MHIS, modified Hachinski ischemic score; SD, standard deviation.
Baseline diagnosis
Plasma VCAM-1 levels for AD, MCI, and CN were 2.89 [2.87, 2.91], 2.86 [2.85, 2.87], and 2.84 [2.82, 2.87] ng/ml (mean [95%CI]), respectively (Fig. 1a). Participants diagnosed with AD had significantly higher mean VCAM-1 levels compared with the MCI (p = 0.0026) and CN (p = 0.0028) diagnostic groups; however, VCAM-1 levels in CN and MCI groups were similar (CN versus MCI; p = 0.2687). Mean ICAM-1 levels for AD, MCI, and CN were 2.04 [2.01, 2.07], 2.00 [1.98, 2.01] and 2.01 [1.97, 2.05] ng/ml. Participants with AD had higher mean ICAM-1 concentrations relative to those with MCI (p = 0.0189), but not in comparison with the CN group (AD versus CN; p = 0.4187) (Fig. 1b). ICAM-1 concentrations were not significantly different in the CN versus MCI group comparison (p = 0.5639). Mean E-Selectin levels for AD, MCI, and CN were 6.56 [6.11, 7.04], 6.58 [6.34, 6.84], and 6.79 [>6.17, 7.47] ng/ml, respectively. E-Selectin levels did not differ between patients with AD, MCI, or CN diagnoses (p = 0.8280, Fig. 1c).
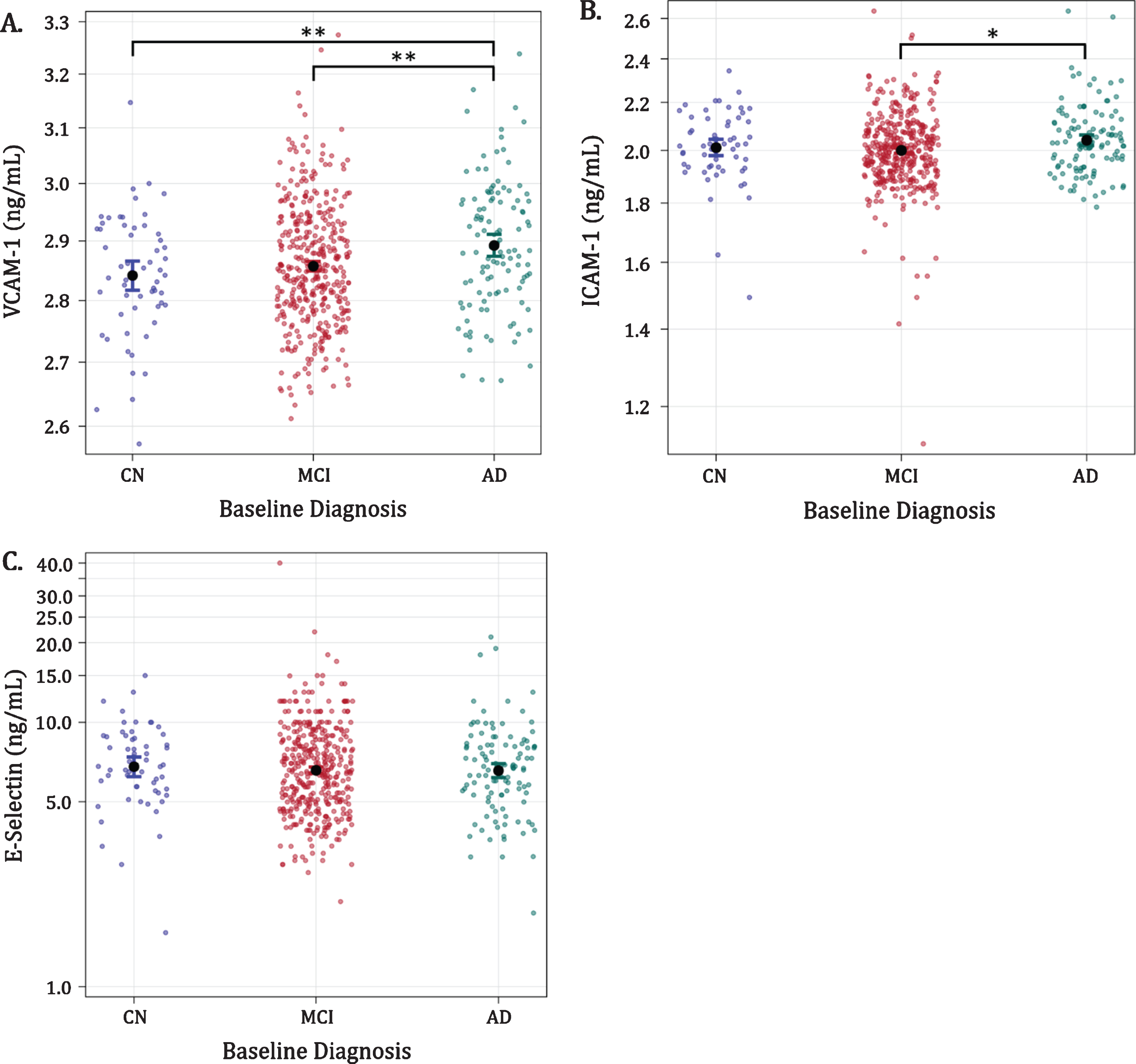
Associations of VCAM-1, ICAM-1, and E-Selectin with baseline diagnosis (figures in logarithmic scale). Dots with bars indicate average levels with confidence intervals. AD, Alzheimer’s disease; ADNI; CN, cognitively normal; ICAM-1, intercellular adhesion molecule-1; MCI, mild cognitive impairment; ng/mL, nanograms per milliliter; VCAM-1, vascular cell adhesion molecule-1; *p < 0.05; **p < 0.005.
Functional status staging and CDR memory sub-score
Across the entire sample, baseline plasma VCAM-1 levels were associated with greater functional impairment stage at study entry as indicated by higher CDR-SB scores (p = 0.0157). Similarly, higher plasma VCAM-1 levels were associated with severity of memory impairment (as indicted by higher CDR-Mem sub-score) at baseline (p = 0.0071). Neither ICAM-1 (p = 0.0645 and p = 0.2489, respectively) nor E-selectin levels (p = 0.5700 and p = 0.6604, respectively) levels were associated with CDR-SB or CDR-Mem scores.
The effects of APOE genotype, age, sex, and family history
Severity of functional status (CDR-SB) modeled by VCAM-1 level and APOE genotype showed that APOE status modified the relationship of VCAM-1 to CDR-SB (Supplementary Fig. 1a). Although participants with higher CDR-SB scores also had higher VCAM-1 levels, regardless of APOE4 status, we observed effect modification by genotype, such that APOE ɛ4 heterozygotes (2.66 [2.39, 2.96]) and homozygotes (3.02 [2.47, 3.69]) (both p < 0.0001) had greater respective functional impairment scores at baseline (e.g., higher CDR-SB scores) than APOE ɛ4 non-carriers (1.68 [1.50, 1.88]).
Age did not significantly influence the association of VCAM-1 with CDR-SB (Supplementary Figure 1b; slope not significantly different than 0, p = 0.1395). Stratifying the sample by sex, we found a trend for female (slope significantly different from 0), but not male sex as a modifier of the relationship between VCAM-1 and CDR-SB (Supplementary Figure 1c; p = 0.1888 and 0.0478 for men versus women, respectively). Family history of AD was also found to modify the relationship of VCAM-1 with CDR-SB. However, this was observed only for positive family history in both parents; this finding was significant before and after adjustment for variance (p = 0.0148 and p = 0.0444, respectively), but there were only 8 participants in this category (Table 1).
As only VCAM-1 was consistently correlated with clinical diagnosis and global functional status, the above analyses were not performed with respect to ICAM-1 or E-Selectin levels.
CSF AD biomarkers
About half of participants also underwent lumbar puncture by ADNI-1 protocol for collection of CSF samples at the baseline visit. For the following analyses, we included a subset of the original sample (n = 351; n = 102 AD, n = 197 MCI, n = 57 CN) with AD biomarker results in CSF (Aβ42, T-tau, P-tau, and P-tau/Aβ42 ratio, log normal distributions), as well as measures of plasma VCAM-1, ICAM-1, and E-Selectin from concurrently collected blood samples.
We found that neither VCAM-1, ICAM-1, nor E-selectin levels across our sample at baseline were significantly associated with any of the CSF biomarkers (Aβ42, T-tau, or P-tau), or with P-tau/Aβ42 ratios (data not shown). Moreover, contrary to our prediction, there was no evidence of interactive association when CAMs were entered with CSF biomarkers into the CDR-SB models individually (data not shown). Instead, we observed significant additive relationships for VCAM-1 when included in CDR-SB models with Aβ42 (p = 0.0022), T-tau (p = 0.0084), P-tau (p = 0.0084), or P-tau/Aβ42 ratios (p = 0.005); we found additive contributions of ICAM-1 to CDR-SB models that included either P-tau (p = 0.0467) or the P-tau/Aβ42 ratio (p = 0.0496) (Table 2).
CDR-SB modeled (additive) by the combination of VCAM-1, ICAM-1, or E-Selectin and CSF biomarkers
Aβ42, amyloid-β 42-residue peptide; CDR-SB, clinical dementia rating-sum of boxes; DF, degrees of freedom; ICAM-1, intercellular adhesion molecule-1; P-tau, phosphorylated tau; T-tau, total tau; VCAM-1, vascular cell adhesion molecule-1.
DISCUSSION
Current epidemiologic evidence suggests that potentially modifiable midlife vascular risk factors specifically influence risk for later-life, AD-ass-ociated cognitive decline. Despite this, a large knowledge gap still exists with respect to characterization of the biological overlap of highly co-morbid vascular and AD pathologies in clinical AD. Given recent emphasis on prevention-oriented strategies in AD, delineating the nature of clinical associations between vascular-mediated processes and AD biomarkers is imperative.
In this study, we first sought to explore whether molecular indicators vascular endothelial dysfunction as indicated by soluble plasma CAM levels would reliably estimate baseline AD-related clinical diagnosis and/or functional impairment in a cohort that was purposefully designed for studying biomarkers in AD, in which contributions from potential confounders such as significant vascular cognitive impairment had been minimized. In our cross-sectional analyses of ADNI-1 data, we found that sample-wide baseline VCAM-1 and ICAM-1 (but not E-Selectin) levels had significant but modest associations with AD-associated diagnosis. We had predicted that CAM levels would be lowest in CN subjects and concomitantly higher in MCI and AD groups, respectively, but found VCAM-1 and ICAM-1 to only be consistently higher in AD versus MCI groups. As an extension of our initial hypothesis, we had predicted that higher baseline peripheral CAMs levels would also correlate with severity of functional impairment staging and found that VCAM-1 (but not ICAM-1 or E-Selectin) again had a significant but relatively modest relationship to CDR-SB scores across the entire sample.
Our main finding was related to our prediction that plasma CAMs and brain-based AD pathology would act synergistically in contributing to clinical severity of AD. Contrary to our initial expectation, we found that plasma VCAM-1 and CSF AD biomarkers acted independently and observed more robust additive effects when added to any of the CSF biomarkers in CDR-SB models. ICAM-1 was also observed to have an additive effect in these models, but to a more limited extent, and we observed no effects with E-Selectin in this model.
Our hypothesis that sensitive molecular indicators of vascular endothelial dysfunction such as soluble plasma CAMs would indicate interactive rather than independent associations of vascular and AD pathologies was largely informed by recent experimental data. Peripheral immune cell trafficking into the brain has been reported in rodent models to occur with specific predilection for brain parenchyma affected by deposition of Aβ [70, 71] and tau [72], and one study reported high levels of both VCAM-1 and ICAM-1 expression in cerebral blood vessels adjacent to Aβ plaques [71]. Moreover, Aβ has been demonstrated to induce a CAM-mediated pro-inflammatory cascade in vascular endothelial cells [73], blood-brain barrier (BBB) dysfunction via endothelial and smooth muscle cell damage [74], and vasoconstriction via free radicals [75]. Interestingly, elevated pro-inflammatory CSF tumor necrosis factor has been shown in a mouse model to drastically increase β-site amyloid precursor protein-cleaving enzyme 1 (BACE1) processing of CSF VCAM-1 from its membrane-bound to soluble form, a process that was not prominent in healthy adult mice [76].
Additionally, Yousef et. al. have demonstrated in a detailed series of studies of aging mice that a) the aged hippocampus expresses an inflammatory transcriptional profile that induces local microglial activation that is spatially associated with focal VCAM-1 upregulation on the luminal side of the adjacent BBB; b) soluble VCAM-1 is elevated in aged mouse (and human) plasma, and aged plasma from mice and humans induces VCAM-1 expression in cultured brain endothelial cells and young mouse hippocampi; and c) the effects of aged blood in this model (including impaired cognition in a Barnes maze) are mitigated by administration of an anti-VCAM-1 antibody or genetic ablation of VCAM-1 [58]. The proinflammatory effects of the dialyzed, aged plasma that was used for experiments were specifically found to not be related to soluble VCAM-1 itself, and authors surmised that pro-inflammatory cytokine/chemokine signaling was likely responsible for the effect [58]. Of note, they found no increased expression of ICAM-1, E-Selectin, or P-Selectin at either mRNA or protein levels in their model [58]. Yousef et al. also observed three distinct populations of vascular endothelial cells, only two of which expressed VCAM-1 together with either pro-inflammatory genes or vascular remodeling and Notch signaling markers [58]. It is interesting to note parallels of these findings with an ApoE -/- mouse model of atherosclerosis, where VCAM-1, ICAM-1, and PECAM were all shown to be differentially expressed and localized in response to hypercholesterolemia; however, only VCAM-1 upregulation showed focal predilection for lesion-prone sites and preceded atherosclerotic lesion formation [41].
Human clinical studies to-date have consistently demonstrated peripheral soluble CAMs to be elevated in relation to various forms of dementia but have yielded inconsistent results as to which CAMs are most clinically relevant. For example, some groups using different combinations of CAMs have shown only VCAM-1 [49], ICAM-1 [51], or ICAM-1 and PECAM-1 [50] to be elevated in AD compared with controls, while Huang et al. found VCAM-1, ICAM-1, and E-Selectin to all be elevated in AD compared with controls, but only VCAM-1 to be associated with dementia severity [48]. With respect to cerebral vascular disease, E-Selectin (but not VCAM-1) was associated with severity of small vessel disease on CT in patients with vascular dementia and AD in one study [49]; however, Huang et. al. found VCAM-1 (but not ICAM-1 or E-Selectin) to be associated with cerebral vascular dysfunction in AD patients as measured by major tract-specific fractional anisotropy quantification [48]. In two cross-sectional examinations of community-dwelling older adults, Tchalla et al. found that elevated plasma VCAM-1 (but not ICAM-1) was associated with cognitive impairment, decline in activities of daily living, slowed gait speed, higher cerebral white matter hyperintensity volume on MRI, and cerebrovascular resistance as measured by transcranial doppler [52, 53]. Another group assessed an expanded panel of neuroinflammatory biomarkers including VCAM-1 and ICAM-1 in serum (and CSF) for cross-sectional associations with CSF AD biomarkers in normal community-dwelling adults and patients with cognitive impairment, and found that eight of the serum (including ICAM-1, but not VCAM-1) biomarkers best predicted a CSF AD profile defined by P-tau/Aβ42 ratio [55]. Longitudinally, the Rotterdam study showed baseline α1-antichymotrypsin, interleukin-6, and C-reactive protein but not VCAM-1 or ICAM-1 to predict future dementia risk [77], while Yoon et. al. reported that soluble VCAM-1, ICAM-1, E-Selectin, and P-Selectin assessed longitudinally in healthy volunteers all increased with age, but only elevated ICAM-1 predicted poorer cognitive performance years later [32]. To our knowledge, ours is the first study to assess for interactive versus additive associations of blood-borne soluble CAMs and CSF AD biomarkers with respect to AD diagnosis and dementia severity staging in a highly characterized AD cohort.
There were several limitations of our study. These included the cross-sectional design, multiple comparisons, and the smaller number of participants with CSF biomarkers relative to the overall cohort size. For simplicity, we specifically chose to limit this initial study to cross-sectional outcomes of AD-associated diagnosis and functional impairment in order to test our basic assumption that elevated CAMs would generally serve as a useful biomarker of vascular contributions to the clinical AD phenotype. Although our data suggest primacy of VCAM-1 among the CAMs tested herein with respect to clinical AD, and parallel rather than interactive associations of soluble plasma VCAM-1 and ICAM-1 to CSF biomarkers of AD-pathology and neurodegeneration, these data should be interpreted with caution in the context of findings from other groups that were generated in studies with differing methodologies as outlined above. It is also possible that available sample sizes and/or the presence of outliers in ADNI data may have influenced our findings related to between-group differences in ICAM-1 and E-Selectin. Additionally, if membrane-bound VCAM-1 is in fact the key factor in mediating vascular-AD pathological interactions across the BBB as suggested by experimental studies, the relative degrees to which individual CAMs are expressed longitudinally, solubilized, and detectable as blood-borne biomarkers in humans with chronic risk factors remains unclear. It may also be that use of blood-borne CAMs in isolation without pro-inflammatory chemokine/cytokine and/or imaging biomarkers is insufficient to fully capture clinically relevant aspects of vascular contributions to AD via mechanisms acting across the BBB.
Despite these limitations, our data lend weight to the growing literature suggesting that soluble blood-borne CAMs may serve as useful biomarkers for studying the biological overlap of vascular and AD pathologies in clinical AD. In order to validate these findings, future studies of CAMs in ADNI data will include assessment of associations with AD and vascular imaging biomarkers, correlations with longitudinal cognitive and functional data, and likely inclusion of CAMs in a more comprehensive panel of peripheral inflammation and BBB dysfunction. Future studies would also benefit from validation across multiple large, longitudinal cohorts using standardized methodologies.
Footnotes
ACKNOWLEDGMENTS
This work was supported by NIA # AG036535 and NIA #AG058648 (Daiello). The funding sources had no role in the design, conduct, or reporting of this study.
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (![]() ). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for NeuroImaging at the University of Southern California.
). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for NeuroImaging at the University of Southern California.