
Abstract
Quantitative modeling is quickly becoming an integral part of biology, due to the ability of mathematical models and computer simulations to generate insights and predict the behavior of living systems. Single-cell models can be incapable or misleading for inferring population dynamics, as they do not consider the interactions between cells via metabolites or physical contact, nor do they consider competition for limited resources such as nutrients or space. Here we examine methods that are commonly used to model and simulate cell populations. First, we cover simple models where analytic solutions are available, and then move on to more complex scenarios where computational methods are required. Overall, we present a summary of mathematical models used to describe cell population dynamics, which may aid future model development and highlights the importance of population modeling in biology.
Introduction
“Let theory guide your observations” - Charles Darwin [1]
Just as a gene seldom functions in isolation from other genes, cells exist as part of a population of cells that affect each other’s function, dynamics, and survival. Cell populations can be genetically homogeneous or heterogeneous, and even in the former case, a significant amount of phenotypic cell-to-cell variability can exist within a population [2–4] and influence fitness [5–8]. Genetic variance has long been known to affect organismal fitness attributable to natural selection through changes in gene frequencies [9, 10], and more recently, nongenetic phenotypic diversity is thought to facilitate evolution [11–14]. Populations of cells generate a myriad of complex behaviors as cells share or compete for resources. There are interesting cases where the short-term fitness of some individuals is sacrificed to “bet-hedge” against future hostile environmental conditions to maximize the long-term fitness of the population [15, 16]. Population-level effects can increase phenotypic diversity via processes such as negative frequency-dependent selection, where rare phenotypes are favored over common ones [17, 18].
While some of these ideas can be investigated using experimentation alone, there are many aspects of these concepts that are hidden from experimental measurement. For example, it is difficult to measure fluctuating intracellular protein concentrations, or the cascade of interactions that occur when the expression of one gene regulates the expression of other genes in the same cell or in other members of the population. The complexity of this process and the difficulty of measuring all aspects of it underpin the necessity of modeling. An experimentalist may be limited in what they can measure (e.g., a handful of fluorescence signals for a few mutants). As a result, they may have several hypotheses which can explain an observed biological phenomenon. By converting these hypotheses into mathematical models, the experimentalist can rigorously test these hypotheses by comparing the models’ predictions against the experimental data (for articles on quantitative modeling in biology see refs. [19–22]). Following Occam’s razor, the simplest model which can predict the data is generally regarded as the best explanation for the biological phenomenon being investigated. As population-level dynamics cannot be captured by modeling a single cell in isolation, we need to develop theoretical and computational population-level models. Population models also make experimentally testable predictions, which narrows down the almost infinite state space of possible wet-lab experiments, and generate insights from the large amounts of data these experiments produce. Some subfields of biology, such as ecology and evolutionary biology, have been adept at utilizing models of population dynamics [23, 24], while other subfields have further to go.
To understand and predict the dynamical behavior of a population of cells, it is imperative to develop models describing the whole cell population and not just a single cell. For instance, it has been shown by several authors that stochasticity in biochemical reactions and unequal partitioning at cell division can lead to complex population dynamics [25–29], the latter of which is not amenable to single-cell models. Furthermore, the growth and evolution of a colony or tumor is often affected by indirect interactions between cells through metabolites or direct physical contact [30, 31], which can only be captured using population models. Another consideration is that in many cases calibrating a model of a single “typical cell” is not possible [32]; though single-cell experimental methods are advancing rapidly [33], at present much of the data are still generated from bulk assays (e.g., western blotting). This is particularly problematic for single-cell models when there is large degree of cell-to-cell variability in the population, as inferring single-cell models from this data can lead to biologically meaningless results [32]. Also, cellular growth dynamics are affected by fluctuating population densities and nutrient availability [34]. In cases such as those outlined above, population-level dynamics differ from those of a single-cell and can generally not be derived or “averaged out” from single-cell observations. Therefore, often the best approach is to develop population models from experimental population data, so that the cell population dynamics are described in a statistically accurate manner.
The literature contains many studies that incorporate cell population models, ranging from differential equation models [35, 36] to complex single-cell agent-based population simulations [37–39], which have led to profound biological insights and guided experiments. For instance, a simple analytic deterministic model of colon cancer cells demonstrated that tumor development is sometimes associated with exponential growth (see Section 2.1) of a population previously at equilibrium, and other times with an increase in cell number at equilibrium as a result of differentiation and apoptosis failure [40]. This model was subsequently refined to incorporate the effect of cell age distribution and mutation to explain long lag phases observed in tumor growth [41]. On the other end of the spectrum, probabilistic models have been used to describe a wide range of phenomena, including stem cell and progenitor population dynamics that arise from cell proliferation and fluctuations in the state of differentiation [42], and the reprogramming of somatic cells into induced pluripotent stem cells [43]. In some cases, models have predicted completely novel phenomena after being fit to existing data, and subsequently verified by population-level experiments (e.g., [44]). These studies are all examples where modeling has led to deeper insights into biological systems, by providing hypotheses that are quantitatively consistent with the observed phenomena.
The focus of this review is on methods that can be employed to quantitatively model and simulate cell population dynamics. For the sake of brevity, we do not discuss spatial models or the theoretical work from classical population genetics. For reviews on spatial models, see refs. [45–47], for a review on molecular population genetics, see ref. [48], and for books on mathematical population genetics, see refs. [49, 50]. We begin by introducing a few simple scenarios that can be solved analytically, which can be useful for describing cell populations while avoiding potentially significant computational complexity [51], and are also useful for benchmarking computer algorithms [52–54]. Computational methods which are required for more complex biological systems are introduced next, followed by a discussion of what is presently lacking and future directions for the field.
Population growth models
Exponential growth
The exponential growth rate of a cell population is commonly used as a measure of “fitness” in experimental microbiology studies (see Section 4.5.1 for a brief discussion of fitness) [14]. This measure of fitness is especially suited for well-controlled single-species experiments, where daily dilutions can be performed to ensure that the cell culture remains in an exponential growth phase. The exponential growth phase is often referred to as the “log phase”, because growth rates are often obtained via linear regression of logarithmic plots of cell growth (which is linear on a log-scale during the exponential growth phase), in addition to fitting an exponential function to linear-scale data obtained experimentally from cell counters, spectrophotometers, and plate readers.
Exponential growth has the advantage of being simple to describe

Modeling cell population growth. (A) Exponential growth, logistic growth, and the Allee effect. (B) Growth curves for the Baranyi model. A single run with no noise [noise strength was set equal to 0 for the numerical solution of Equation (13); red solid line] and ten independent runs of the Baranyi model with noise [noise strength was set equal to 0.035 in the numerical solution of Equation (13); blue dashed lines]. The Matlab codes and parameters used to generate (A) and (B) are available at: https://github.com/dacharle42/MCPD_ISB (Color online).
The major limitation of modeling growth as an exponential process is that the exponential phase of growth is short-lived in biologically realistic scenarios, as exponential growth is only one phase of growth for cell populations (see Section 2.2). Another limitation is that natural populations of single species do not often exist in isolation and it is unclear how exponential growth relates to competition assays (see Section 4.5.1).
Logistic growth models were first established by Verhulst in 1838 [55]. Since then, logistic-type models have been widely applied to topics ranging from the role of autophagy in yeast cell population dynamics in response to starvation [56] to evolutionary studies of cancer [57]. As with the exponential growth model, the logistic model describes how population size changes in time
Equation (3) can be derived from the following set of coupled ordinary differential equations (ODEs)
Substituting Equation (7) into Equation (4) we obtain
Factoring the above equation yields
The logistic equation [Equation (3)] can be solved analytically to obtain the population size as a function of time
Note that in the case where the number of cells in the population is small compared to the environmental carrying capacity (N ⪡ K), we recover from Equations (3) and (10) exponential growth [Equations (1) and (2), respectively, noting that since we are modeling exponential growth that N0 ≤ N and thus N0 ⪡ K]; that is, exponential growth is a special case of the more general logistic growth model. Unlike the exponential model, which only has a trivial (unstable) fixed point at zero, the logistic model also has a stable fixed point at K (Fig. 1A). Eventually, N will approach the value of K, which denotes the size of the population at the stationary phase where the net population growth rate is zero. This contrasts with exponential growth, in which the population size tends towards infinity, showing that on longer timescales logistic growth is more realistic. Logistic growth models do not account for cell death, which for instance can occur from a lack of nutrients, high concentrations of accumulated toxic wastes, or other stressful conditions. This is the final portion of the standard growth curve, characterized by an exponential decrease in N. The cell-death phase, where some cells loose viability or are destroyed by lysis, is avoided by properly timed cell culture dilutions but still occurs in certain experimental conditions and natural settings that are stressful to the cell population.
The Monod model is a widely used mathematical model that relates population growth rate r to the concentration of a limiting resource [58, 59]. The Monod equation is
The Allee effect, a biological phenomenon where the size of the population affects individual growth, is a common deviation from logistic growth [57, 61]. Allee effects are generally applied in ecology to mating populations, but have also been incorporated into models of cancerous cell populations [62]. A strong Allee effect describes a population that can grow at intermediate population densities but declines when the number of organisms is either too small or too large (i.e., per-capita growth rate reaches a maximum at intermediate population size). A weak Allee effect is where the population growth rate is small but positive for small N, but without a critical population size or density under which the growth rate becomes negative. Populations that are subject to Allee effects can collapse and become extinct if their population size falls below the critical threshold; this poses a challenge for conservation biologists but provides a therapeutic avenue for oncologists [57]. The strong Allee effect where growth rate is negative at small N is described by the following ODE
Unlike the exponential and logistic growth equations, an exact explicit solution does not exist for the Allee effect equation [Equation (12)] and therefore a solution must be obtained numerically.
Lag-time (or adaptation time) is one critical aspect of the growth curve that is not well captured by the models presented in Sections 2.1–2.4. For example, lag-time optimization has been shown to contribute to antibiotic tolerance in evolved bacterial populations [63]. The Baranyi model accurately describes the lag-phase and transition to exponential phase and takes the form [64, 65]
The Baranyi model can be applied to any of the models presented so far in Section 2 to describe the transition period before exponential growth or decay. The physiological state of individual cells is affected by their previous growth environment and exposure to stressful conditions, which can extend the lag phase and increase cell-to-cell lag-time variability [63, 74]. Other models have been used to determine adaptation time, such as the lag-exponential and Gompertz models [64, 75]. However, fits to the same data yield similar lag-time values as the Baranyi model, which is less influenced by the quality of the dataset and provides the best fit in the majority of cases [64, 76].
In this section we present an example that demonstrates the importance of modeling cell populations and how population growth models can be developed to predict or interpret experimental results. We include the relevant steps required to develop such a model for those less familiar with quantitative modeling; several of the steps also highlight the utility of mathematical modeling for identifying quantitative relationships between biological variables that can be measured experimentally.
Consider a cell population where some cells randomly arrest growth due to non-optimal environmental conditions. Two subpopulations will emerge: actively growing cells (C) and arrested cells (A). The population dynamics can be described by the following set of “reactions”
This implies the following growth equation for the total number of cells N,
Importantly, we can conclude from Equations (18) – (20) that the apparent growth rate of the whole population g must be different (smaller) than the growth rate of the active cells. Our goal will be to estimate g
C
and g
r
from g, C, and A (quantities that can easily be measured experimentally). The first equation is solvable analytically
Substituting Equation (21) into Equation (19) gives the number of inactive cells
Therefore, the total number of cells is given by
The exponential growth of the whole population can also be described by Equation (21) which implies
The long-term limit (t→ ∞) yields
Therefore, the growth rate of the active cells is equal to the sum of the arrest rate and the growth rate of the whole population. The fraction of arrested cells will be given by the ratio
From here, we obtain another relationship between g r and g c , namely
Finally, we find for g c
Likewise, the arrest rate g r will be given by
This simple example highlights the utility of mathematical modeling for extracting quantitative relationships from the biological systems that can be verified experimentally. For instance, the growth rate of the entire population (g) can be obtained by using an automatic cell counter or a spectrophotometer to measure the optical density of the cell culture. The growth rates of the active subpopulation (g c ) and the arrest rates (g r ) can be determined from image analysis of single-cell time-lapse microscopy data [77]. These experimental measurements can be used to validate predictions made by the model or serve as input to the model to make novel predictions to be validated by further experimentation.
Markov modeling has been used to infer and annotate morphological state and multiphenotype properties from experimental data [78, 79]. The following is a simple illustrative example of applying a Markov chain model to mother-bud Saccharomyces cerevisiae yeast pair transitions among four different phenotypic green fluorescent protein (GFP) reporter expression states (Fig. 2A) in a stressful high-temperature environment. In the transition matrix [Equation (31)], P ij is the probability that, if the mother-bud GFP state is i (row), then it will be followed by state j (column). The columns (left to right) correspond to the states S11 (GFP expressing mother-bud cells), S10 (GFP expressing mother cell and non-expressing bud cell), S01 (non-expressing mother cell and expressing bud cell), and S00 (non-expressing mother-bud cells), as do the rows (top to bottom) in the same order. The transition matrix is given by
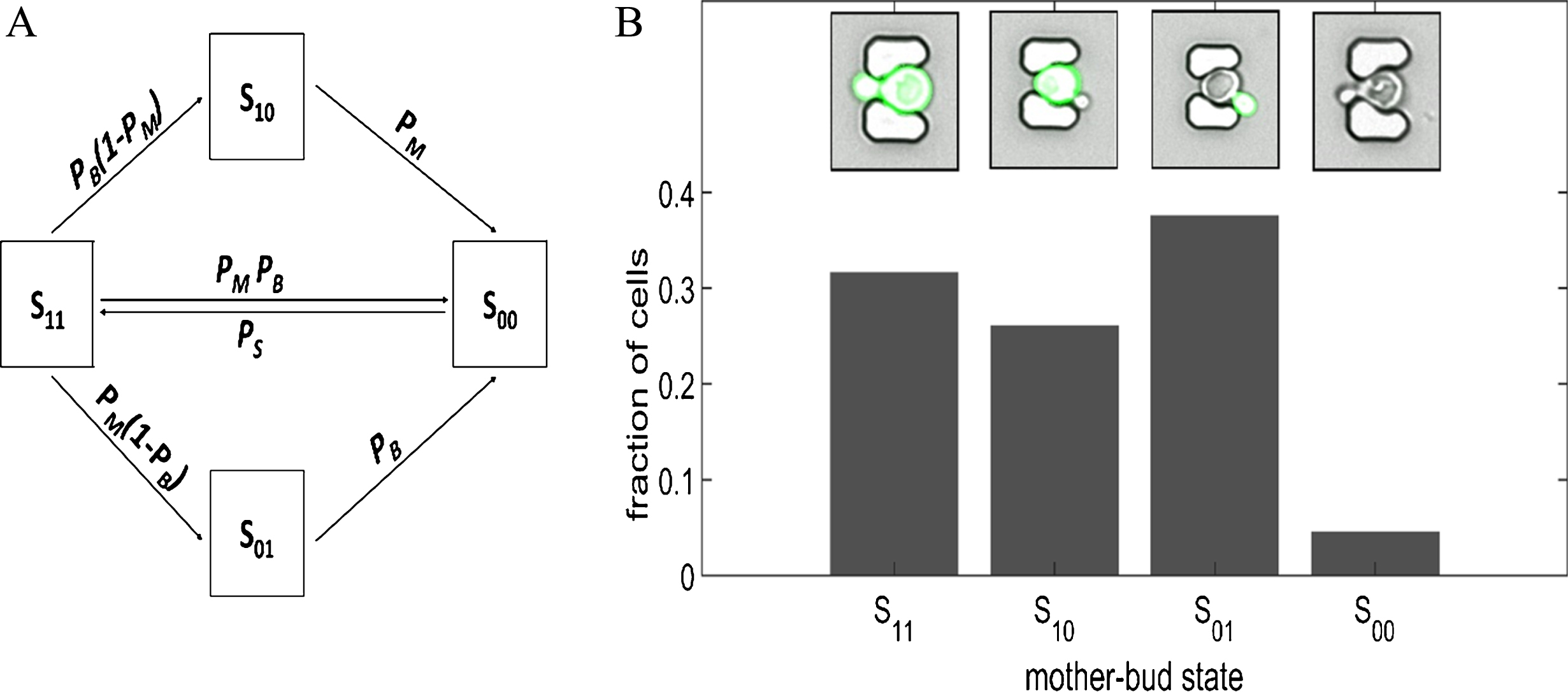
Markov chain model of mother-bud GFP expression states in a Saccharomyces cerevisiae budding yeast cell population exposed to high-temperature stress. (A) Schematic of phenotypic expression states and possible mother-bud state transitions that form the bases of the Markov chain model. Mother-bud pairs in cultured at high temperature (38°C) could be in one of four possible states: S11 (GFP expressing mother-bud cells), S10 (GFP expressing mother and arrested non-expressing bud), S01 (non-expressing mother and resistant expressing bud), S00 (non-expressing mother-bud cells). (B) Population fractions of mother-bud states generated from the Markov chain model. Insets are microscopy images of budding yeast cells at 38°C, obtained in our lab using a modified high-throughput yeast aging analysis (HYAA) microfluidics chip [33], of the possible mother-bud states for the Markov chain model exemplar. The Matlab code and parameters used to generate (B) is available at: https://github.com/dacharle42/MCPD_ISB (Color online).
where P M and P B are the probabilities that the mother and bud cells shut off GFP expression, respectively, P D is the probability that an arrested non-expressing mother-bud pair will remain in the microfluidics trap, and P S is the probability that the mother-bud pair will start expressing GFP. In this example, some of the cells attempt to survive the heat stress by autophagy [80]. Here it is assumed that only non-expressing mother-bud pairs can leave the microfluidics trap as a result of autophagic cell death (the cells shrink when they die and fall through the trap) due to persistent environmental stress [81].
The steady-state fraction of mother-bud pairs in each phenotypic state can be predicted by
Population balance equations
Quantitative modeling plays an important role in bridging the relationship between single-cell and cell-population dynamics. The origins and consequences of cell-to-cell variability are often investigated analytically or computationally using single-cell models (e.g., [53, 91]). Such models typically ignore or idealize cell division and rarely capture the population-level effects of differential reproduction. An approach known as population balance modeling addresses this using partial differential equations [25, 92–97]. In this approach, cells are described as a continuous density flowing through a multi-dimensional state space that quantifies different physiological attributes (e.g., mass and chemical composition). Integral terms are used to account for birth and death processes along with a function describing the partitioning of cell contents at division. In the simplest one-dimensional case with no nutrient limitation or cell death, the population balance equation (PBE) can be formulated as follows
where F (x, t) is the number of cells, x is the cell mass, r (x) is the growth rate function of x, γ(x) is the division rate function that describes how the probability of a cell division varies with x, and is a partition function that describes the probability of a cell that divides with mass x to produce two sibling cells with masses x′ and x-x′. The first term on the left-hand side of Equation (33) is a transient term and the second is an advection term; the right-hand side of the equation contains the source and sink terms.
The time-evolution is uniquely determined by the initial population distribution and all cell densities change according to the same deterministic rules. Correspondingly, information about individual cell trajectories is lost in this formalism. PBE models can be extended to include growth dependence on an external substrate and a diffusion term can be added to account for randomness in the evolution of cells in the state space. The modeling of discrete morphological stages or phases of the cell cycle can be represented by a set of coupled partial differential equations. Most PBEs cannot be solved analytically but can be discretized and integrated numerically. Unfortunately, when more details and higher dimensionality are incorporated, population balance models quickly become very difficult to formulate and solve computationally [25].
ODE models typically use first-order decay terms to account for dilution due to exponential growth [2]. Similarly, discrete stochastic models approximate the effect of growth and partitioning of cell contents at division by increasing the degradation rates of all components. These methods tend to average away the dynamics resulting from growth and division, which can play an important role in cell dynamics (e.g., stochastic partitioning at cell division [98], asynchronously dividing cells [99], and asymmetric division [100–102]). In fact, it has been suggested by Huh et al. [98] that much of the cell-to-cell variability that has been attributed to gene expression “noise” (expression differences between genetically identical cells in the same environment) instead comes from random segregation at cell division, due to the difficulty in separating partitioning errors and gene expression noise profiles. Incorporating the details of cell division and gene expression into population models may help to resolve such controversies.
Computationally, cell growth and division can be modeled explicitly. Recent single-cell experiments show that the cell volume increases exponentially in bacteria [103–106], yeast [106–110], and mammalian cells [106, 111], and can be modeled using an exponential function [52, 112–114]
If cells grow at a rate that is proportional to the amount of protein they contain [115, 116], and if the protein concentration is constant, cells will grow exponentially in mass and volume [117]. Modeling growth at the cellular level (see Section 4.4) can be important, as variability in single-cell growth rates may decrease the population growth rate [118]. Such models for volume changes can be used to better capture intracellular protein dilution rates, since the dilution rate of any intracellular protein is given by
If V (t) happens to depend on other cell types, then we can model dilution rates more generally using the above formula.
In addition to considering how the cell grows, one must also consider, based on the biology of the cell, how a cell regulates its size and how the cell volume and contents will be distributed at division. Cell size homeostasis may occur through different modes of regulation, including “sizers”, “adders”, and “timers”. Sizer regulation requires that a cell monitors its own size and cell division does not proceed until a minimal size has been reached [119]. Adder regulation occurs when a cell adds a fixed size increment before division. Similarly, timer regulation describes the case when a cell grows for a fixed time duration before division. These three main modes of cell size regulation can be captured by the following equation [117]
Variability in size at division, or “sloppy cell-size control”, can be incorporated by treating cell division as a random process that takes place with a volume-dependent probability [121]. Asymmetry in cell division can be modeled in either of these cases by setting the sum of the sibling cell volumes (V1 and V2) equal to the total volume of the dividing cell (V)
Models of non-interacting and non-dividing cells have been used extensively to study population variability arising from the process of gene expression. The population statistics from these models are often calculated from simulation of numerous independent realizations of individual cells [124, 125]. To incorporate cell division into the population’s dynamics, one might propose two approaches to investigate how populations evolve in time:
Simulate the time courses of single cells, randomly choosing one of the two newborn cells to follow when a cell divides. The result is lineages (or cell chains) containing a single individual per generation (e.g., [53]). Simulate the time courses of single cells and continue to simulate all newborn cells produced. The result is a complete lineage tree (e.g., [122, 126]).
One problem with using the cell chain approach is that it does not account for the proliferative competition between cells in different physiological states, and so fails to provide the correct joint distribution of cell properties except in special cases. Hence, the second approach must be used when dealing with a model in which cell proliferation can vary with a number of intrinsic variables, such as age, metabolic state, and cell type. The problem here is that the size of the simulation ensemble rapidly grows to the point of intractability. This can be addressed using a Monte Carlo technique called the constant-number Monte Carlo (CNMC) method [127–129], originally developed to approximate the solution of population balance models (Section 4.1) of particulate processes.
The CNMC method is a statistically accurate method that keeps the total number of cells in an exponentially growing population fixed by randomly selecting cells at a user-specified time interval (which corresponds to experimental cell culture dilution times). This method is particularly suited to modeling well-mixed liquid cell cultures and has been applied to simulate heterogeneous cell populations [26, 130]. The Gillespie algorithm [131, 132], which allows exact individual-based simulation of stochastic mass action kinetics, was combined with the CNMC method to accurately and efficiently simulate gene expression dynamics across growing and dividing cell populations [52].
Population dynamics algorithms
Population dynamics algorithms (PDAs) are computational frameworks that are important for studying cell population dynamics because they can account for a wide range of phenomena that cannot be investigated analytically or by simulating a model of a single cell. PDAs accomplish this by simulating a sufficiently large ensemble of individual cells, serving as a representative sample of the “true” population. This is advantageous because it puts the available methods for single-cell simulation at our disposal without the difficulty of integrating complicated and heterogeneous single-cell behavior into a broader mathematical framework. Notably, the use of individual-based models places virtually no constraints on the biologically relevant details that can be formulated and simulated [133]. For instance, this allows biologically realistic features to be modeled with relative ease, such as cell growth and division effects (Section 4.2), that can be difficult to formulate and solve in an analytical framework.
Previous studies [123, 134] paved the way for the individual-cell based PDAs. Though many of these previous approaches are extremely useful, they can be computationally prohibitive for simulating the dynamics of large, exponentially-growing cell populations. Motivated by this limitation, algorithms for the simulation of intracellular content and cell growth and division were developed, ranging from deterministic [130] and stochastic Langevin approaches [26], along with methods to determine the timing of cell divisions and partitioning of cell contents that agree with population balance formulism (Section 4.1), to parallel stochastic approaches that described growing and dividing cells [52, 114]. One method, which we refer to here as the “asynchronous PDA”, combines the Gillespie stochastic simulation algorithm [131, 132] with a CNMC method (Section 4.3) to simulate population dynamics in a computationally efficient manner (Fig. 3A). Here, “asynchronous” refers to cells being simulated independently from each other and the global population state being determined (and statistics calculated) at discrete and usually equally spaced synchronization barriers to permit parallelization of the algorithm. In the asynchronous PDA, the population is restored at the synchronization barriers to a predefined size using the CNMC method. An accelerated version of the asynchronous PDA, which is applicable when steady-state and symmetric cell division can be assumed, was subsequently developed [114]. The accelerated PDA is well suited for expediting simulations by performing coarse-grained explorations of parameter space, to be subsequently investigated in more detail using the asynchronous PDA.
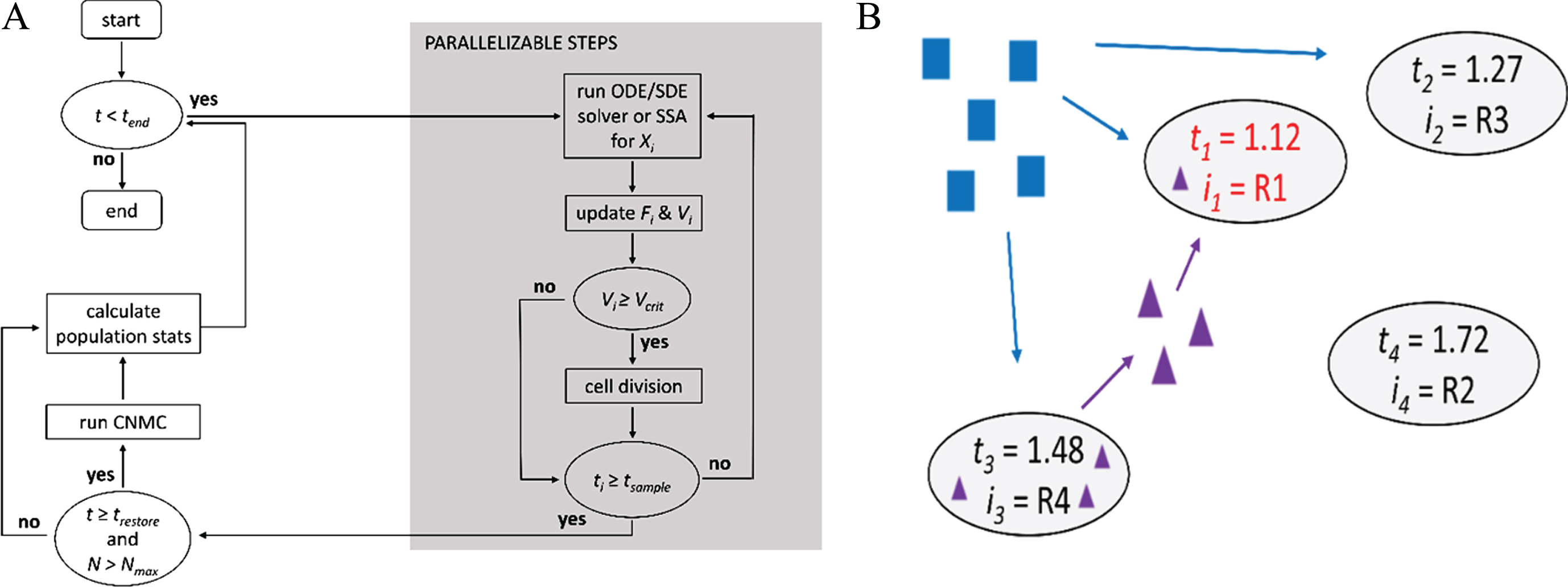
Population dynamics algorithms. (A) Flow diagram for the asynchronous population algorithm, where all cells are simulated independently of one another and synchronized only when the simulation time for each cell (t i ) is equal to or exceeds the user specified sampling time (t sample ). The population size is restored using a constant-number Monte Carlo (CNMC) method to a prespecified fixed size each time the simulation time (t) is greater or equal to the population restore time (t restore ). Xi is the system of equations/reactions and Fi the fitness that correspond to cell i, respectively. (B) Schematic illustrating the concept of a general population simulation framework. Which reaction occurs next (R i ) and the time at which it will occur (t i ) in each cell is determined stochastically. This approach allows for intracellular communication (represented by purple triangles and arrows) and resource consumption (represented by blue squares and arrows) in “real-time” [as opposed to only at each t sample in (A)]. Here, the next reaction will occur for cell 1 (i1 = R1) at t1 = 1.12, when it will uptake a signaling molecule exported from cell 3 at an earlier time. Fortran code for (A) is available in the Appendix B of [176] and at: https://github.com/dacharle/PDA_Fortran, and an object-oriented C++ prototype at: https://github.com/alanyuchenhou/gene-expression (Color online).
Another more general framework, inspired by the concept of “reaction channels” in Gillespie’s algorithm [131, 132], links simulation channels through scheduling dependency graphs (introduced by Gibson et al. [135] to improve the performance of the Gillespie algorithm) to handle the scheduling and execution of state-updating events on individual cells [136]. This approach is analogous to Gillespie’s algorithm, where the propensities of different reaction channels are used to determine when the next reaction will occur (scheduling) and how the numbers of molecules are changed when it occurs (execution) (Fig. 3B). Simulations are performed using an asynchronous method, which is ideal and parallelizable for non-interacting cells, and a synchronous method, enabling the incorporation of cell-to-cell communication (which is not practical in the PDAs due to the parallel nature of the algorithms). For a discussion of synchronous versus asynchronous models, see ref. [20]. Once the population size limit is reached, the CNMC method (Section 4.3) is introduced (as in the asynchronous PDA) to keep a fixed sample population size with the appropriate composition.
In summary, the dynamics of heterogeneous cell populations can be highly complex and are difficult to investigate analytically. The frameworks presented in this section address this by enabling efficient individual-based population-level simulations without the need to formulate or solve complex mathematical equations. They are designed specifically to ease the incorporation of user-designed biological features and to facilitate the transition towards population-level modeling in quantitative biology.
Many of the mathematical models and computer algorithms discussed so far in this review article can be modified to model evolution. This is important, for example, because it allows us to perform long-term in silico evolution experiments in scenarios that may be difficult or costly to investigate in the laboratory. In Section 4.5.2 we present a simple computational model of evolution; more complex computational frameworks to model the evolution of a cell population are discussed in Section 4.5.3.
Fitness
Darwin’s theory of evolution by natural selection is built on the idea that some genotypes have higher fitness than others. However, what exactly mean by the term “fitness” is not always clear and term has been used to mean subtly different things [48]. In fact, even the unit of selection, whether it be the gene or individual [137, 138] or group (recently rebranded as multilevel selection theory) [139, 140], is still debated. A related concept is inclusive fitness, which describes the total effect an individual has on proliferating its genes by producing off spring and by providing aid that enables relatives to reproduce [141]. An evolutionary strategy for increasing inclusive fitness, even at a cost to the individual’s own survival and reproduction, is known as kin selection. According to Hamilton’s rule, kin selection causes genes to increase in frequency when the genetic relatedness of a recipient to an actor multiplied by the benefit to the recipient is greater than the reproductive cost to the actor. Fitness landscapes have long been used to illustrate the effect of genetic factors on fitness [142], and more recently, nongenetic factors as well [8, 13].
The exponential growth rate (Section 2.1) is one common measure of fitness in microbiology and experimental evolution studies. At the population level, this is done by measuring the number of cells or the optical density of the cell culture (e.g., using a cell counter or a spectrophotometer, respectively) and then obtaining the growth rate by fitting the data to an exponential function [Equation (2)]. Population fitness is also commonly measured by direct head-to-head competition assays [143]. This is often done experimentally by determining the relative fitness of each competitor with respect to a reference strain. For example, non-fluorescing evolved cells can be competed (and distinguished) against an ancestral strain that expresses the green fluorescent protein [144]. The fitness (W) is then calculated by
Cell growth rates within a clonal population can vary depending on the environmental context in which it evolved. While a constant environment selects for low variance in growth rate, a fluctuating environment can select for high variance if the growth rate correlates with survival under stress [85]. Thus, the growth-rate distribution is an important evolutionary parameter that can be captured using single-cell experimental measurement techniques [146] and modeled using population simulation algorithms (Section 4.4).
Population fitness is distinct from the cellular fitness of its constituent members, though the former can be obtained from the latter. For instance, we can define the population or “macroscopic” fitness W of an isogenic population under stress as [120]
The model presented in this section describes how the number of cells with wild-type and mutant genotypes varies over time based on their fitness [14]. Specifically, this model describes population dynamics by a system of ordinary differential equations and assumes a constant population size and mutation rate. Here, wild-type and mutant cells are characterized by a single fitness parameter. This ODE evolution model (a corresponding but more detailed evolutionary simulation framework is discussed in Section 4.5.3) has three free parameters: rate of beneficial mutations μ, input probabilities P(G) and P(T) of a given mutation being type G (genomic) or, K (knockout) or T (tweaking); P(K) is determined via P (K) = 1 - P (G) - P (T). Note that while the probability of P(K) could be 1, its rate μP (K) is ⪡1 per genome per generation.
The approach taken in this model is similar to that presented in Section 4.1, where the “gain” and “loss” rates of each genotype are used to develop a system of ODEs that describes the population size of each genotype i over time. Assuming that the number of beneficial mutations arising per unit time is proportional to the number of cell divisions (i.e., mutations arise strictly due to DNA replication errors), it can be described by
If we consider only the influx of new genotypes that survive drift (i.e., assuming all other mutations go extinct rapidly), then the effective influx of genotypes M
i
that carry a potentially beneficial mutation of type i equals
This model can now be applied to specific cell types by defining the ancestral/mutant types (M0/M i ) and the associated fitnesses (f0/f i ). This modeling approach is more general than the more detailed computational approach that is briefly discussed in the following section and facilitates large-scale parameter scans.
The ODE evolution model predicted how fast experimental wild-type genotype disappears from the population, as well as the mutation type (K, T, G) that predominantly replaces the wild type in each condition [14]. Interestingly, the ancestor genotype disappeared fastest in conditions with steep monotone cell fitness landscapes (see [149] for a review of fitness landscapes) and remained in the population longer in peaked cell fitness landscapes; each experimental condition favored different fractions of mutations types as long as they were available.
More detailed computational evolutionary simulation frameworks to model molecular evolution have been developed that explicitly account for experimental details (such as phenotypic switching and resuspensions). One such framework from the same study [14] as the ODE evolution model described in Section 4.5.2 enables the prediction of how experimental evolution will affect evolutionary dynamics (Python code is available in the supplemental materials of Gonzalez et al. [14]). A more detailed computational framework was required because the simpler ODE evolution model could not predict the number of distinct mutant alleles in the evolving population and lacked important experimental details (e.g., periodic resuspensions and phenotype switching between ON and OFF states with experimentally determined switching rates). The evolutionary simulation framework predicted the number of distinct mutant alleles, in addition to the characteristics predicted by the simpler mathematical model. Importantly, the modeling in Gonzalez et al. [14] was crucial for understanding the evolutionary dynamics of mutants arising, establishing, and competing, as well as the number of alleles in the cell population.
Another computational framework that can be used to model the evolution of cell populations is the asynchronous PDA (Section 4.4), which was recently modified to incorporate evolution [13]. This was done by modeling genetic mutation as a change in the reaction rate parameters of a mutant cell probabilistically each time a cell divides. As cell fitness (cell cycle time) is coupled to gene expression level and selection pressures, this allows for selection of the most fit genotype. Importantly, this approach provides a framework for studying how nongenetic variability in gene expression can affect evolution and predicted that the level of evolved cell-to-cell variability in the population depends on the associated fitness costs and benefits of gene expression in a specific environment. An exact algorithm for fast stochastic simulations of evolutionary dynamics was developed by Mather et al., [150], which provides a significant speedup when the population size is large and mutation rates are much smaller than the birth and death rates.
Discussion
We have covered some of the common mathematical and computational methods for investigating cell population dynamics. To balance comprehensiveness with length of the review, several modeling approaches were omitted out of necessity, not due to lack of importance. For instance, spatial models can be critical for predicting the behavior and fate of cell populations. Spatial models often involve compartmentalized ordinary differential equations [35], stochastic differential equations of motion [38], partial differential equations and various computational approaches that have been derived from cellular automata [45]. These models have been used to describe a wide range of phenomena, from T cell population dynamics [35], to tumor growth, cancer metastasis, and chemotherapy resistance [151]. For cases where only “population snapshot” data are available, which provide single-cell measurements at every time point (such as with flow cytometric analysis) but do not provide single-cell time series data (because the cells are discarded after each measurement), Bayesian approaches can be used to identify models of heterogeneous cell populations [32]. In a complementary fashion, there are also methods available to deconvolve cell population dynamics from single-cell data (e.g., [152]).
Model parameterization is as important as the structure of the model. Much effort has been devoted to developing optimization techniques, which involve scanning the parameter space to minimize a cost function (i.e., minimize the error between the output of the model and the experimental data) [153]. Common optimization techniques include linear and nonlinear least-squares fitting [154], simulated annealing [155], genetic algorithms [156], and evolutionary computation [157, 158]. The problems with optimization methods are that they can be computationally expensive and may not perform well on noisy data sets [153]. Bayesian methods can infer whole probability distributions of the parameters (rather than just a point estimate) when the data includes measurement noise and/or intrinsic noise. The challenge here is that analytic approaches are generally intractable and numerical solutions are challenging due to the need to solve high-dimensional integration problems. Maximum likelihood estimation has also been widely applied [153, 160]. The appropriate parameter inference method depends on the modeling framework, and moving between frameworks (e.g., deterministic to stochastic) may involve recalculating the parameters. For example, parameters for zero order (e.g.,
The field of computational biology is presently lacking comprehensive “user-friendly”, customizable, multiscale, computationally efficient cell population simulators, though many of these individual components are available in different software packages. CellPD, a user-friendly open source software, was developed for experimental biologists (without specialized training in computational or mathematical modeling) to automatically quantify key parameters of cell phenotypes based on fits of various mathematical cell population dynamics models to the experimental data [34]. One limitation of CellPD is that it does not support user-defined mathematical models without modifying the source code (for Python source code and executable files see ref. [165]). As mathematical modeling gained traction in biology, simulators with graphical user interfaces (GUIs) were developed to aid biologists [38]. One example is CellSys, a modular software tool developed for off-lattice simulation of growth and organization processes in multi-cellular systems in 2D and 3D [38]. To try and offset the major performance bottleneck (solving the stochastic equations of motion for each cell), the core algorithms in CellSys are parallelized using OpenMP (openmp.org), as in the asynchronous PDA. The Open Systems Pharmacology Suite is an excellent example of a software platform for multiscale modeling and simulation of whole-body physiology, disease biology, and molecular reactions networks, which facilitates efficient model development, simulation and model analysis across multiple physiological scales [166]. This software platform combines GUI-based tools (PK-Sim and MoBi [167]) with interfaces to computing environments (R [168] and Matlab [169]) for solving ordinary differential and delay differential equations. Another example is the Glazier-Graner-Hogeweg based CompuCell3D simulation environment, though Python scripting or C++ coding is required to develop modules for implementing customized or more complex models [38].
Future directions in the field involve extending existing software or creating new platforms that biologists can easily use to formulate and simulate spatial and stochastic models of cell population dynamics. The backend of such a simulator could take advantage of distributed-or shared-memory architectures [52], as well as high performance graphical processing units [170]. Machine-learning approaches are another promising direction to take advantage of increasingly large experimental data sets to build multiscale biological models [171], and to bridge the gap between detailed descriptions of intracellular molecular events [124, 125] and population dynamics. Though much progress has been made by studying single or isolated pairs of populations, true multiscale population models will one day have to account for ecological dynamics that result from interactions between a diverse set of populations [172–175], as well as intra-population dynamics in fluctuating or spatially structured environments.
The utility of quantitative models is to gain insight into living systems and make experimentally verifiable predictions. Due to the multiscale nature and combinatorial complexity of biological systems, it is difficult to develop general models that always apply. Until we gain a better understanding of the systems we are modeling, and computational resources and processing times become more ideal, we will be constrained to make approximate and somewhat ad-hoc models. Nevertheless, by testing various models and their ability to predict the experimental data, we can rigorously verify hypotheses about biological phenomena by comparing model predictions to experiments, and in an iterative cycle, improve our models based on the data. These models will continue to serve as a “microscope”, allowing us to peer deeper into nature than experimental methods at the time allow.
Footnotes
Acknowledgments
The authors would like to thank Tamás Székely Jr. for help acquiring the microfluidics time-lapse images, Matthew Wu for assisting with the literature review and comments on the manuscript, Mirna Kheir for help with the literature review, and Teresa Charlebois for editing the manuscript. We would like to give a special thanks to Michael Cortes for insightful discussions and helpful comments on manuscript. This work was supported by an NVIDIA Corporation Titan Xp GPU grant to D.C, an NIH/NIGMS MIRA grant (R35GM122561) to G.B., and the Laufer Center for Physical and Quantitative Biology.