
Abstract
A promoter is a brief stretch of DNA (100–1,000 bp) where RNA polymerase starts to transcribe a gene. A DNA (Deoxyribonucleic Acid) base pair is a fundamental unit of DNA structure and represents the pairing of two complementary nucleotide bases within the DNA double helix. The four DNA nucleotide bases are adenine (A), thymine (T), cytosine (C), and guanine (G). DNA base pairs are the building blocks of the DNA molecule, and their complementary pairing is central to the storage and transmission of genetic information in all living organisms. Normally, a promoter is found at the 5
Introduction
The promoter, a section of DNA, is where RNA polymerase binds to start a gene’s transcription process. According to Lin et al. (2018), promoter sequences are commonly found right before or at the 5
Deoxyribonucleic acid, sometimes known as DNA, is the special genetic code that is responsible for the coordination and functioning of all living beings. DNA consists a double-helix polymer, a spiral consisting of two DNA strands wound around each other [1]. The DNA of almost every cell in a person’s body is identical. The four chemical bases adenine (A), guanine (G), cytosine (C), and thymine (T) make up the DNA code. Each base also has a sugar and phosphate molecule connected to it; this grouping of bases, sugars, and phosphates is known as a nucleotide. The genome of the organism is made up of the set of chromosomes that make up the DNA within each cell. The majority of the DNA in eukaryotes that is not expressed in an amino acid chain of a protein is referred to as “junk DNA”. It is known that only 1.5% of human genome is to be protein-coding [2]. Only one copy of the DNA from the parent organism is present in each cell, and it is located in the nucleus. Therefore, the cell creates a copy of that specific code segment called ribonucleic acid (RNA) inside its nucleus in order to use that particular component of the DNA code.
The promoter region of the DNA plays a key regulatory role in gene expression. A promoter is a section of DNA that designates a gene’s transcriptional start site. An enzyme known as RNA polymerase II binds to a promoter to carry out transcription shown in Fig. 1, in which DNA is converted into RNA, which is then spliced into an mRNA, which is then translated into a protein.
Promoters differ at their consensus sequence according on the RNA polymerase’s factor used, which provides DNA recognition specificity [11, 12]. The plethora of experimentally known promoter sequences for several growth factors in various organisms has made it possible to apply prediction approaches based on the construction of Position-Weight Matrices (PWM), which uncover conserved canonical motifs. The high frequency of false positive predictions with partial coverage, however, limits the effectiveness of these approaches [13, 15].
The present study used a dataset with 106 DNA sequences, each with 57 consecutive nucleotides, from the UCI Machine Learning Repository. In this study, the problem of identifying transcription sites, which entails identifying promoter sequences in short DNA sequences of Escherichia coli bacterium, is resolved. The precise prediction of promoter sites is essential for comprehending gene expression, deciphering patterns, and constructing genetic regulatory networks because promoters are now so crucial for transcription.
Transcription process [8].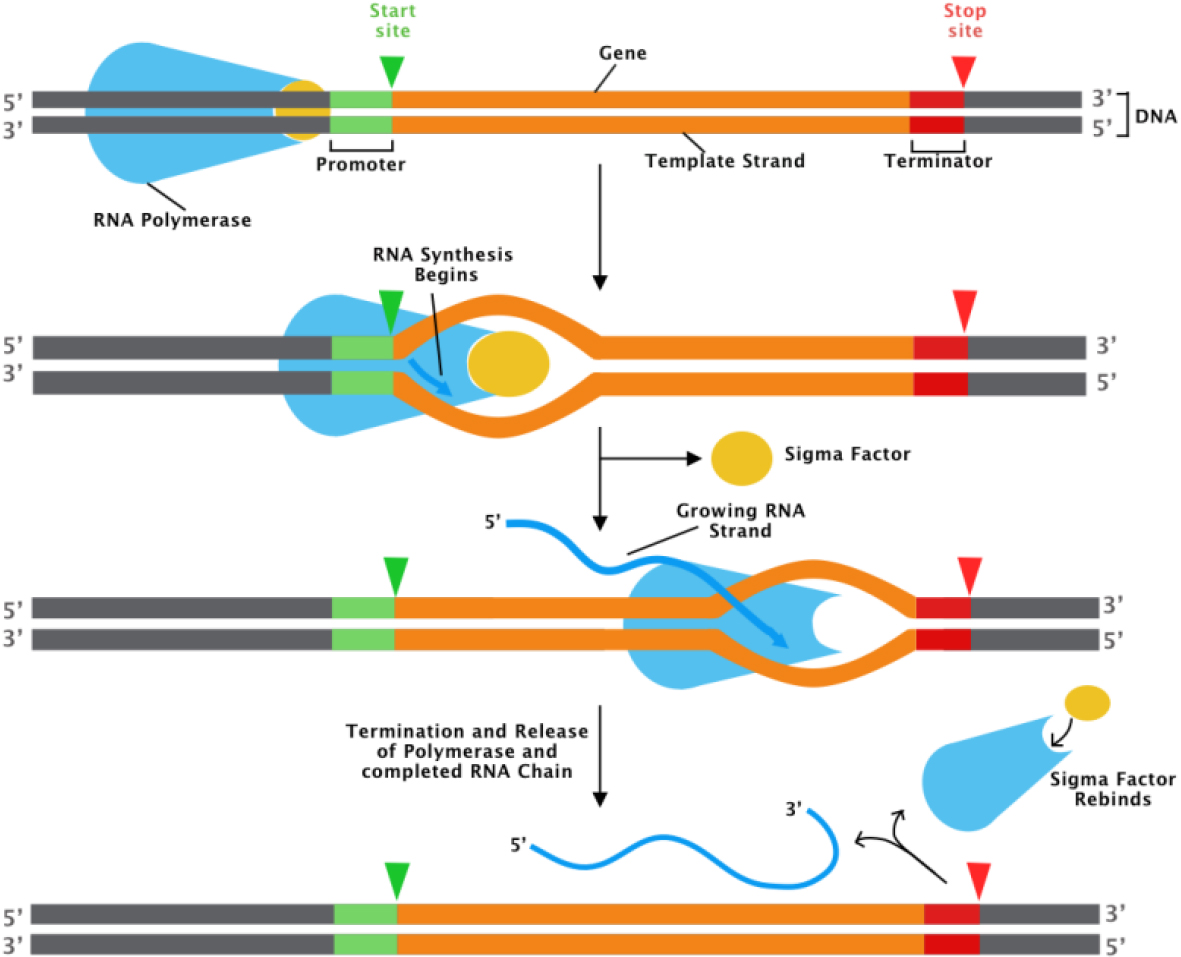
In addition to this, promoter prediction is a crucial step in the genome annotation process because it allows for the identification of novel genes, particularly those linked to non-coding RNA, which are frequently overlooked by gene prediction algorithms. Although numerous intricate feature extraction strategies and numerous classifiers have been put out so far for promoter recognition, the issue is still unresolved [17]. A significant barrier to bioinformatics’ genome-wide investigation of gene regulation is the inability to predict promoters [18].
The database of E. coli experimentally determined promoters has been significantly increased by our extensive promoter identification work, which will encourage experimental biologists to investigate the gene expression mechanisms underlying some of these genes, particularly for those without a known biological function.
Here, the Gaussian Decision Boundary Estimation in machine learning models is proposed to classify the promoters based on the DNA sequence.In order to maximise the performance of the models, the best features are determined using a score-based algorithm to choose pertinent nucleotides that are directly responsible for promoter recognition. With the help of these features, the support-vector-machine model, which is based on Gaussian Decision Boundary Estimation, is trained to identify the optimal hyperplane for classifying the data.
Numerous attempts have been made to recognise promoters using computational methods. Basic Local Alignment Search Tool (BLAST) is a search technique that Altschul et al. [3] proposed as a method for determining the similarity between two genetic sequences. Towel et al. [4] proposed the KBANN (Knowledge-Based Artificial Neural Networks)hybrid learning system. This system blends explanation-based learning with empirical learning for promoter prediction. Using multilayer perceptron neural networks, Weinert et al. [5] describe a quick and effective biomolecular classification methodology that is used to categorise proteins and infer their functions by comparing their structural similarity. Gordon et al. [22] developed an approach for prediction of promoters and Transcription Start Sites in E. coli, based on an ensemble of Support Vector Machines and this classifier is then combined with Position Weight Matrices. It is concluded that an ensemble-SVM with mismatch string kernels may be able to identify and take use of a variety of regulatory motifs for improved TSS/promoter detection.
To predict promoter sequences in the DNA of E. coli bacteria, Tavares et al. [6] examined the efficacy of various machine learning algorithms. In this comparative investigation, results from probabilistic methods like the Hidden Markov Model (HMM) and Bayesian methods are more precise. ANN shave has proven acceptable profound results, but the high false positive rate [20, 21] has affected the specificity. Kemal Polat et al. [10] proposed a novel method based on feature selection (FS) and Artificial Immune Recognition System (AIRS) with Fuzzy resource allocation mechanism (Fuzzy-AIRS) for identifying promoters in strings that represent nucleotides.
Karlı G et al. [9] developed a new method known as IREM (Inductive Rule Extraction Method) for solving the problem. Attribute-value pairs with higher information value is determined by IREM. It uses its own “cost function” to determine how much information each pair in the set is worth. It discussed the notion that a lower cost is a sign of a higher information value. Attribute-values with higher values are given a higher priority while creating the rule base of the forecasting system.
Anveshrithaa et al. [7] observed that promoters in DNA sequences are best classified using neural networks trained with back-propagation. The back-propagation optimisation and grid search hyperparameter tuning are credited with the excellent accuracy. It is discovered that additional models, such as the ensemble learning technique Bootstrap Aggregation and the Support Vector Machine with linear kernel, are as good in categorising DNA sequences. The comparison of the various methods applied to the task of identifying promoters in DNA sequence is shown in Table 1.
Khan z, Rajdeep et al. [8] claimed an improved accuracy by selective choice of hyperparameters for Multilayer Perceptron (MLP) Neural Network in Promoter Identification. Numerous alternative computational techniques have also been put forth. However, the vast majority of these methods do not result in promoter recognition accuracy rates that are satisfactory.
The Inductive Rule Extraction approach is introduced by Karl et al. [9], while the Fuzzy-AIRS classifier and feature selection are used by Polat et al. [10] to identify the DNA sequence’s promoters. For the classification of the promoters from the DNA sequence, Towel et al. [4] and Weinert et al. [5] implement Knowledge-Based Artificial Neural Networks and Multilayer Perceptron Neural Networks, respectively.
Comparison of proposed model with other state of art algorithms
Comparison of proposed model with other state of art algorithms
An overall flowchart of the strategy used is shown in Fig. 2, which operates using a specially created pipeline. In the subsections that follow, each experimental step of this suggested pipeline will be discussed in turn.
The main contribution of the work is listed here,
The dataset required for identification of promoters in DNA sequences is collected and analysed from the UCI Repository. It contained a set of 106 DNA sequences and their corresponding classes (Promoter or Non-Promoter). The DNA sequences are split into 57 individual nucleotides each and are converted to numerical data through one hot encoding by replacing each categorical column in the DataFrame with a set of binary columns which is followed by transformation of the dataset into polynomial feature matrix of degree 2. The ANOVA F-value for each feature in the feature matrix is computed and the top 1000 features are extracted for training and testing. The width of the gaussian function is determined by computing the gamma ( The optimal hyperplane that maximizes the margin between the classes in the transformed feature space are found using the pairwise similarities.
Proposed architecture.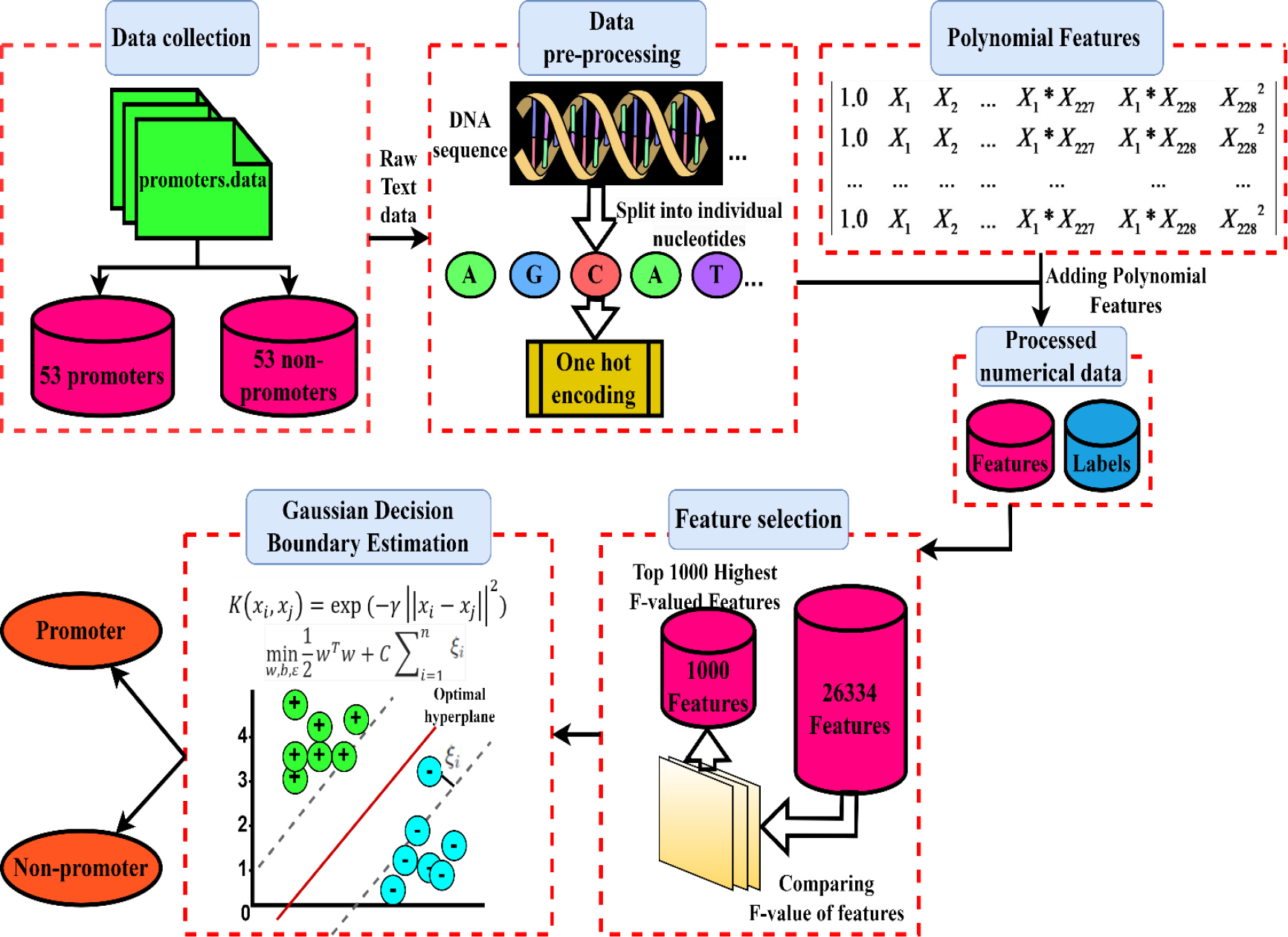
One of the most crucial phases in solving a bioinformatics issue is gathering a high-quality dataset. To unbiasedly compare the performance of our model to other ones already in use, we used the benchmark dataset from the UCI repository in this study.
The dataset for the proposed study is found in the UCI Machine Learning Repository (
Hierarchy of the DNA nucleotides.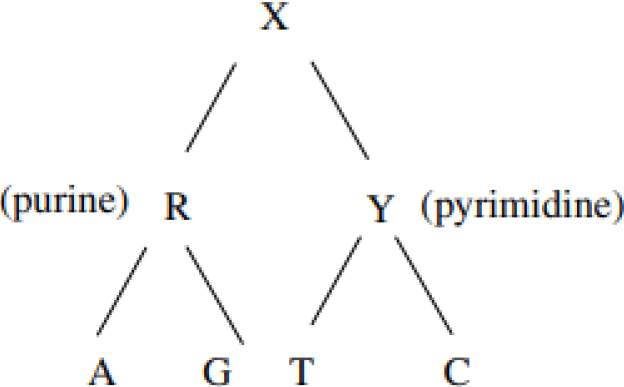
DNA nucleotides can be categorized into two distinct groups: purines, which encompass adenine and guanine, and pyrimidines, which encompass cytosine and thymine in the context of DNA, and uracil when considering RNA. This categorization is predicated upon the structural characteristics of their respective nitrogenous bases.
Purines: Purines constitute a class of nitrogenous bases present within the DNA molecule, characterized by their dual-ring structure. Within DNA, there exist two primary purine bases:
Adenine (A): Adenine represents one of the quartet of nitrogenous bases forming the DNA structure. It engages in the formation of hydrogen bonds with thymine (T) in complementary base pairs within the DNA double helix. In the context of RNA, adenine instead pairs with uracil (U). Guanine (G): Guanine, another purine base present in DNA, pairs with cytosine (C) through the mediation of hydrogen bonds. Notably, guanine also maintains its pairing with cytosine in RNA structures. Pyrimidines: Pyrimidines, the other class of nitrogenous bases within DNA, possess a singular-ring structure. Within DNA, three pyrimidine bases are identified:
Cytosine (C): Cytosine, a pyrimidine base, pairs with guanine in DNA through hydrogen bonding interactions. Remarkably, cytosine also exhibits this pairing behaviour with guanine in the realm of RNA. Thymine (T): Thymine, as another pyrimidine base in DNA, forms specific hydrogen bond interactions with adenine. However, it is essential to note that in RNA, thymine is replaced by uracil (U), which similarly pairs with adenine. Uracil (U): Uracil, exclusive to RNA, represents a pyrimidine base. In RNA molecules, uracil engages in hydrogen bond-based pairing with adenine, mirroring the role of thymine in DNA.
The DNA sequences in the dataset are divided into two classifications, “
The dataset contains a balanced number of samples for each class. A balanced dataset, where each class has an equal number of samples, helps prevent class imbalance issues. Class imbalance can bias the model towards the majority class, making it less sensitive to the minority class. Also, a balanced dataset allows for a more accurate assessment of the model’s classification performance.
The UCI Repository’s data is not in a format that may be used. Prior to training various machine learning models, datasets are pre-processed. Individual nucleotides are separated into the DNA sequences. The numerical data is to be converted from the textual data corresponding to the different nucleotides. One-hot encoding is accomplished for the conversion. One-hot encoding is a technique used to convert categorical data into a binary vector representation [24]. One-hot encoding preserves all the information in the categorical variable. One-hot encoding was used rather than other methods as many encoding methods (e.g., label encoding) assign integer values to categories, which could introduce a magnitude bias. Models might misinterpret the magnitude of these values as meaningful information. One-hot encoding eliminates this issue by using binary values. One-hot encoding also prevents biasing the model towards the category with higher integer values, ensuring that all categories have equal weight. A binary column isgenerated for each categorical column (A, C, G, T and
Polynomial features are generated from the set of encoded features. The encoded input feature matrix
A new feature matrix is created from the original feature matrixand the dataset is transformed into polynomial features. In accordance with the calculation above, each of the 106 DNA sequences will be equipped with 26335 features in the updated feature matrix
Where,
Feature selection using
score function
Feature extraction is a pivotal step in bioinformatics problem-solving, wherein primary features play a crucial role in distinguishing DNA sequences. The abundance of attributes and occurrences in raw datasets often poses significant challenges for data mining systems. A proven remedy to address this issue is feature selection, which aims to identify a compact subset of relevant features while retaining the original purpose through justifiable criteria. By eliminating irrelevant and redundant features, this process results in a simplified dataset, yielding more concise and comprehensible outcomes. The top 1000 features from the feature matrix are selected based on the ‘f_classif’ scoring function and the training data is fitted. The SelectKBest function in the scikit-learn package is used to accomplish this [16].
The f_classif function computes the ANOVA (analysis of variance) F-value between each feature and the target variable. ANOVAor Analysis of Variance, is a statistical technique used to compare means of two or more groups to determine if there are significant differences between them. It helps in understanding whether a categorical factor has a statistically significant impact on a continuous outcome variable. The ANOVA F-value is a statistical measure used to compare the means of two or more groups. It assesses whether the differences in means among these groups are likely due to real differences in the population or random variation. A high F-value, along with a small p-value, suggests significant differences among group means, indicating that the factor being tested has a statistically significant effect on the dependent variable. The higher the F-value, the more likely the feature is to be relevant for classification. A new feature matrix is created containing the top 1000 features determined by the F-value. The proposed method utilizes ANOVA as a set of parametric statistical models and associated estimation techniques to assess whether the means of multiple data samples originate from the same distribution. Each feature is subjected to a univariate statistical test, as it is compared to the target feature, facilitating the identification of statistically significant associations [25].
The F-valueis computed as follows:
Where,
A higher between-group variance signifies a more pronounced distinction between the classes, implying increased relevance of the feature for classification purposes. To identify the most pertinent attributes for classification, the selection of the top 1000 features based on their F-valuesis employed, favouring those with the greatest F-values.
Gaussian decision boundary estimation
Support Vector Machines are a class of supervised machine learning models employed for tasks involving classification and regression. These models aim to construct a hyperplane (as illustrated in Fig. 3) within a high-dimensional feature space, with the primary objective of maximizing the separation between the two classes denoted by “
The Radial Basis Function (RBF) kernel, also known as the Gaussian kernel, stands out as a prominent choice for evaluating the similarity between two samples. The RBF kernel is defined as:
Where,
The number of features taken is 1000, thus
The input data are transformed by the RBF kernel into a higher dimensional space where they are more linearly separated. The optimization problem can be formulated as:
subject to:
Where,
SVM Hyperplane.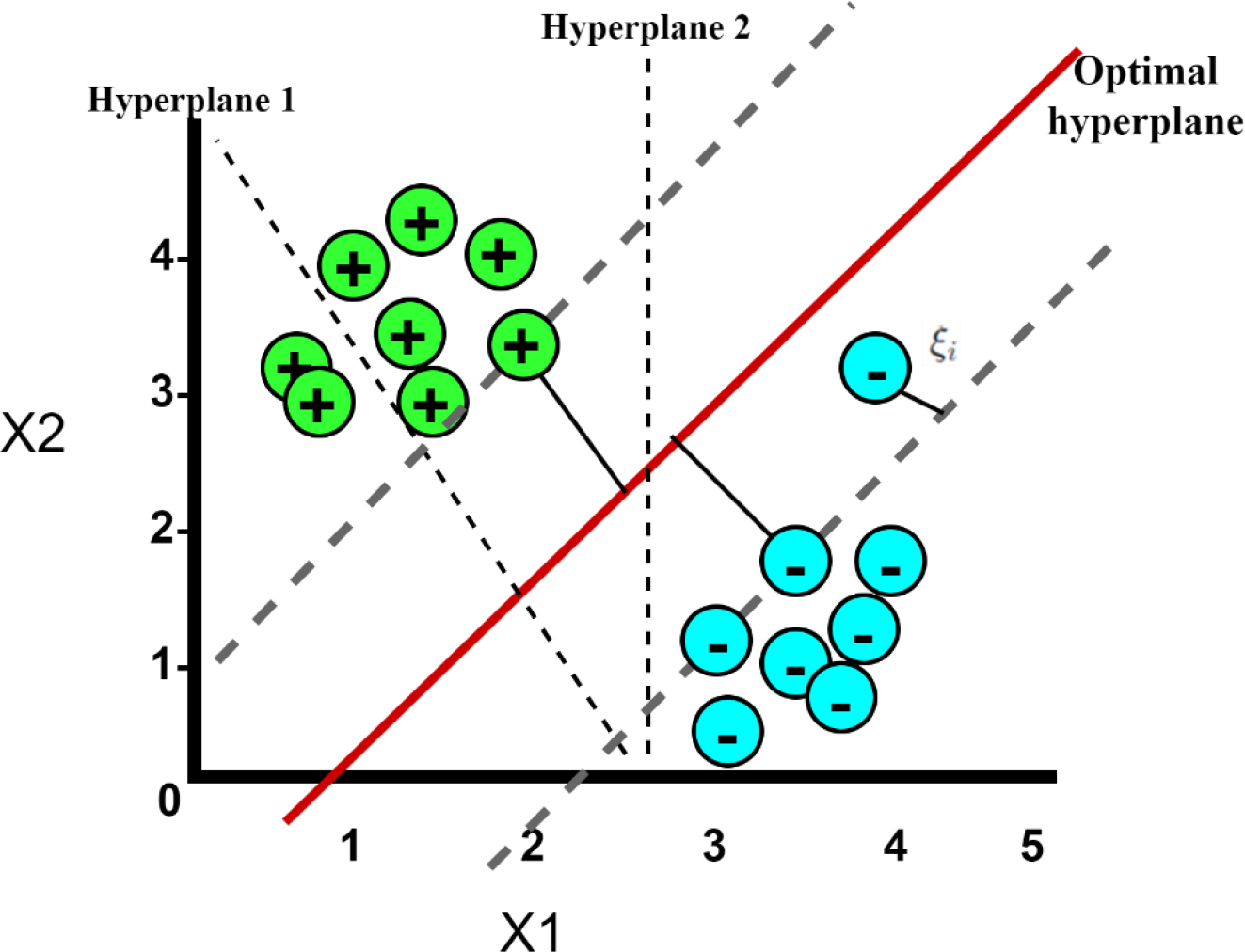
The slack variable represents the amount by which data points are allowed to violate the margin and the classification boundary. They quantify the degree of misclassification for each data point and represents how far a data point is from being correctly classified. The slack variable transforms the hard constraints of perfectly classifying all data points, which might not be feasible, into soft constraints, making the model more flexible and adaptable to complex datasets. It plays a significant role in finding a balance between maximizing the margin and minimizing classification errors, thereby making the model effective for a wide range of classification tasks.
The decision function has the following equation after the Lagrange duality approach yields the dual version of this optimisation problem:
Where,
The Gaussian Decision Boundary estimator is trained on the transformed and selected training data. For every pair of samples
Frequency of classes (1-promoter, 0- non-promoter).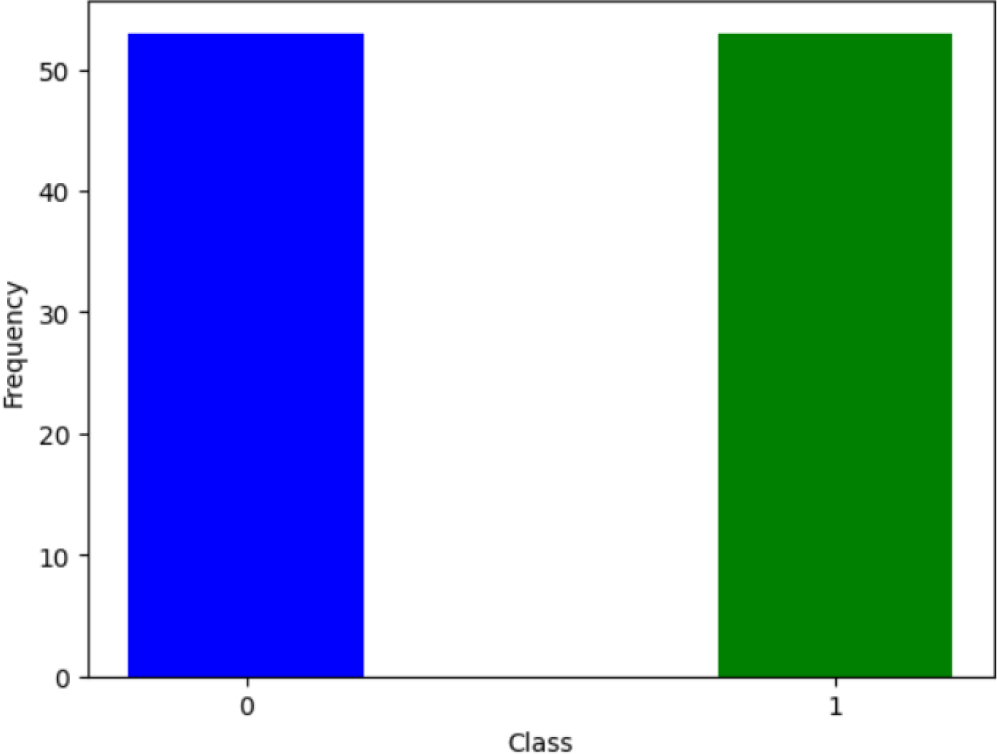
This research endeavour is executed using Python version 3.10.10. For the classification of DNA sequences, the Scikit-learn library, a comprehensive Python machine learning package offering diverse classification, regression, and clustering techniques, is employed. In addition to that, the NumPy and Pandas libraries are utilized for dataset preprocessing. The versions of the libraries utilized are Numpy: 1.23.5, SKlearn:1.2.2 and Pandas:1.5.3.
The PolynomialFeatures in scikit-learn was the tool used for generating new features by creating polynomial combinations of existing numerical features. It was useful for capturing the non-linear relationships in data. SelectKBest is a feature selection technique provided by scikit-learn (sklearn.feature_selection). It selected the top k most important features from the dataset based on a scoring function. f_classif is one of the scoring functions available for this purpose, and it’s specifically designed for classification tasks. The classification report from scikit-learn was significant because it provides a concise summary of key classification metrics for a machine learning model. It included crucial information such as precision, recall, F1-score, and support for each class.
Dataset collection and processing
At the outset, the primary dataset encompassed 106 DNA sequences, accompanied by their respective textual class labels denoted as either positive (
Feature correlation matrix.
The computation of scores for each of the 26,334 features is performed through the utilization of the
Score of each feature using f_classif function.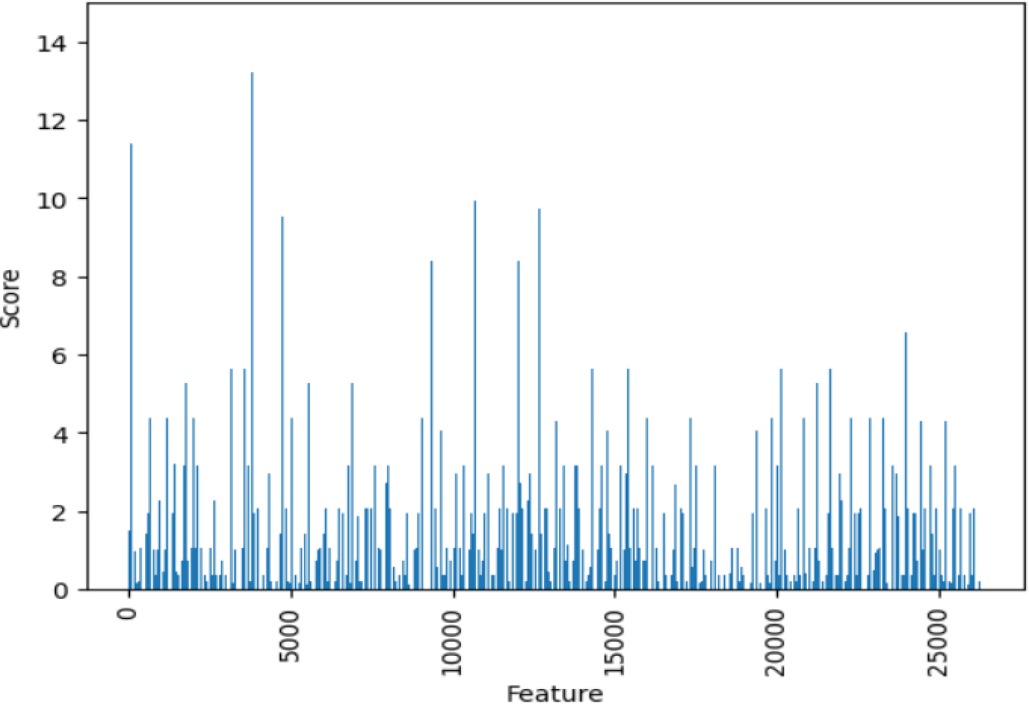
Selected features (Top 1000).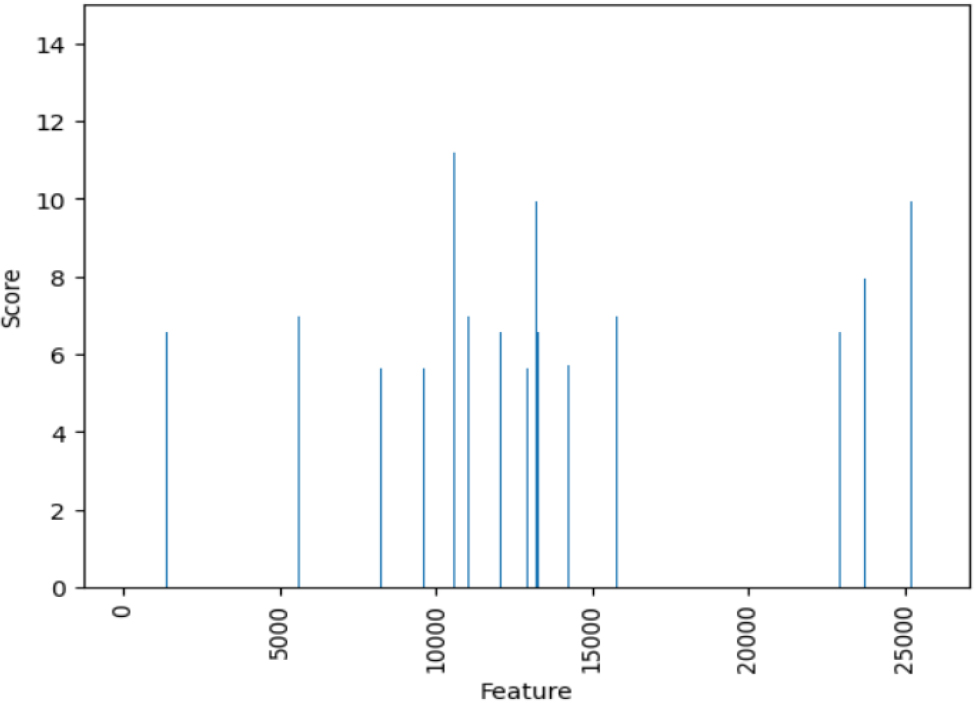
The proposed study employs the Gaussian Decision Boundary Estimator, with the Regularization parameter (C) set to 1.0. Following the training of the Gaussian Decision Boundary Estimator using the selected features, it is subjected to testing. The test dataset comprises 32 sequences, with an equal distribution of 16 promoters and 16 non-promoters. A comparative evaluation is conducted against various alternative machine learning models, including K-Nearest Neighbours (KNN), Gaussian Process Classifier, Decision Tree, Random Forest, Multilayer Perceptron (MLP), AdaBoost, and Naïve Bayes. Notably, the proposed model demonstrated superior performance, achieving an accuracy of 99.9%, as visually depicted in Fig. 10.
Classification report.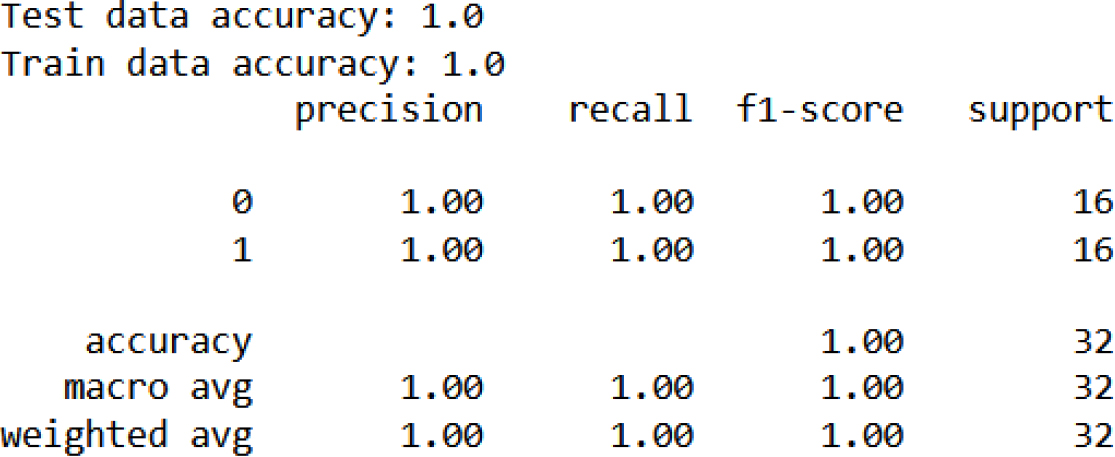
Confusion Matrix and ROC curve for the proposed model, KNN, MLP, Gaussian, Naïve Bayes, SVM (Linear kernel) and AdaBoost.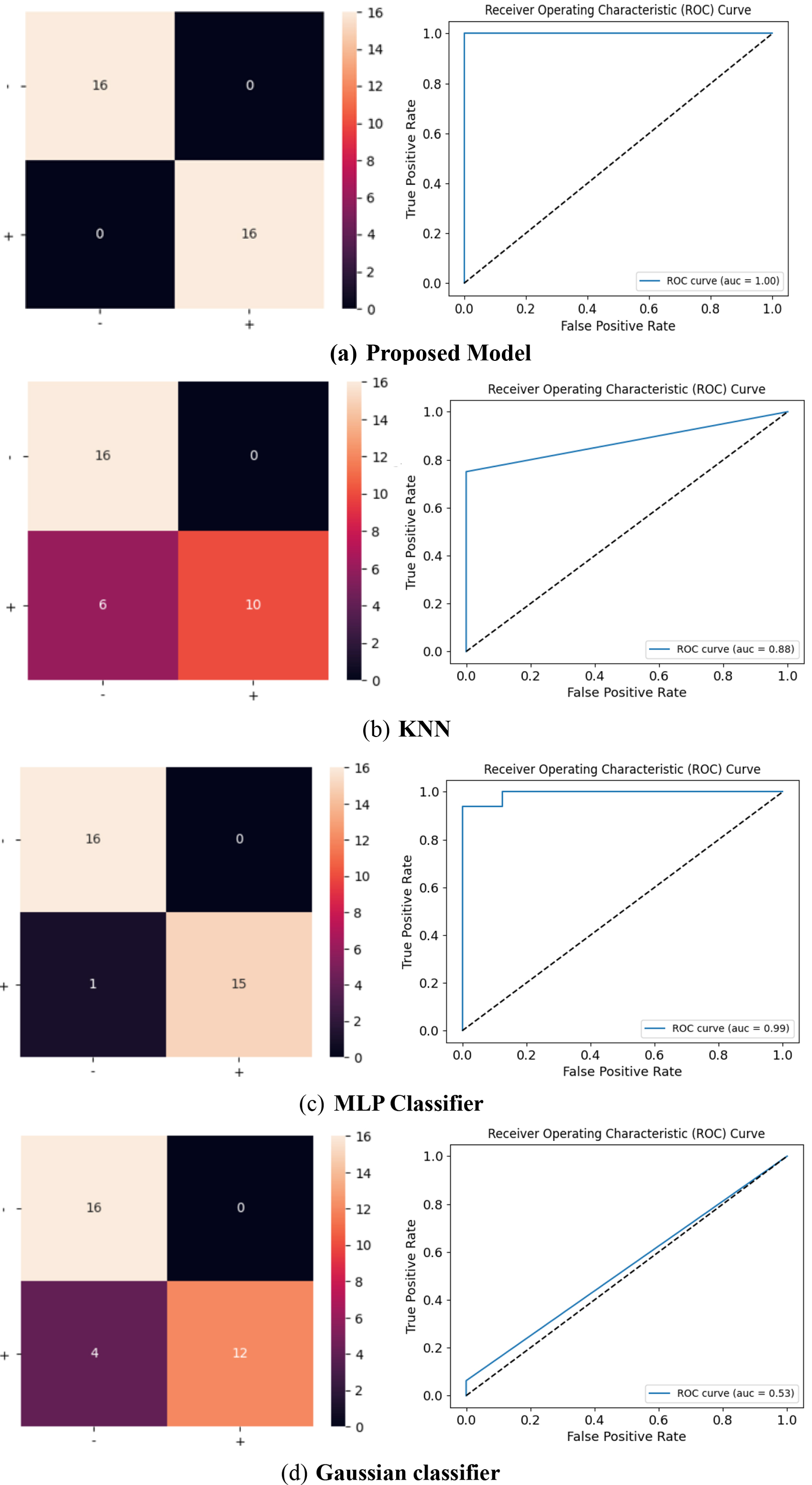
continued.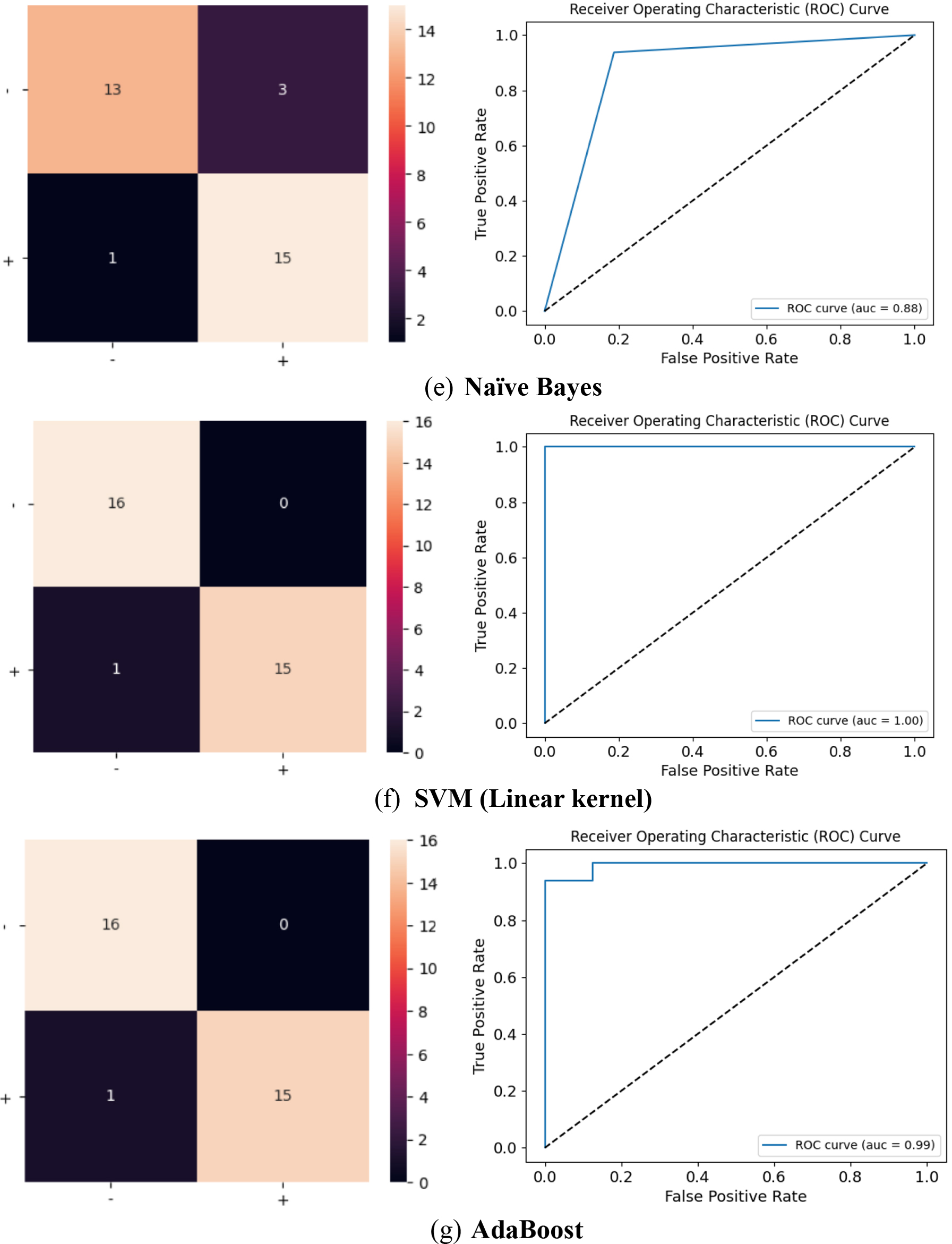
Comparison of the proposed algorithm with other models.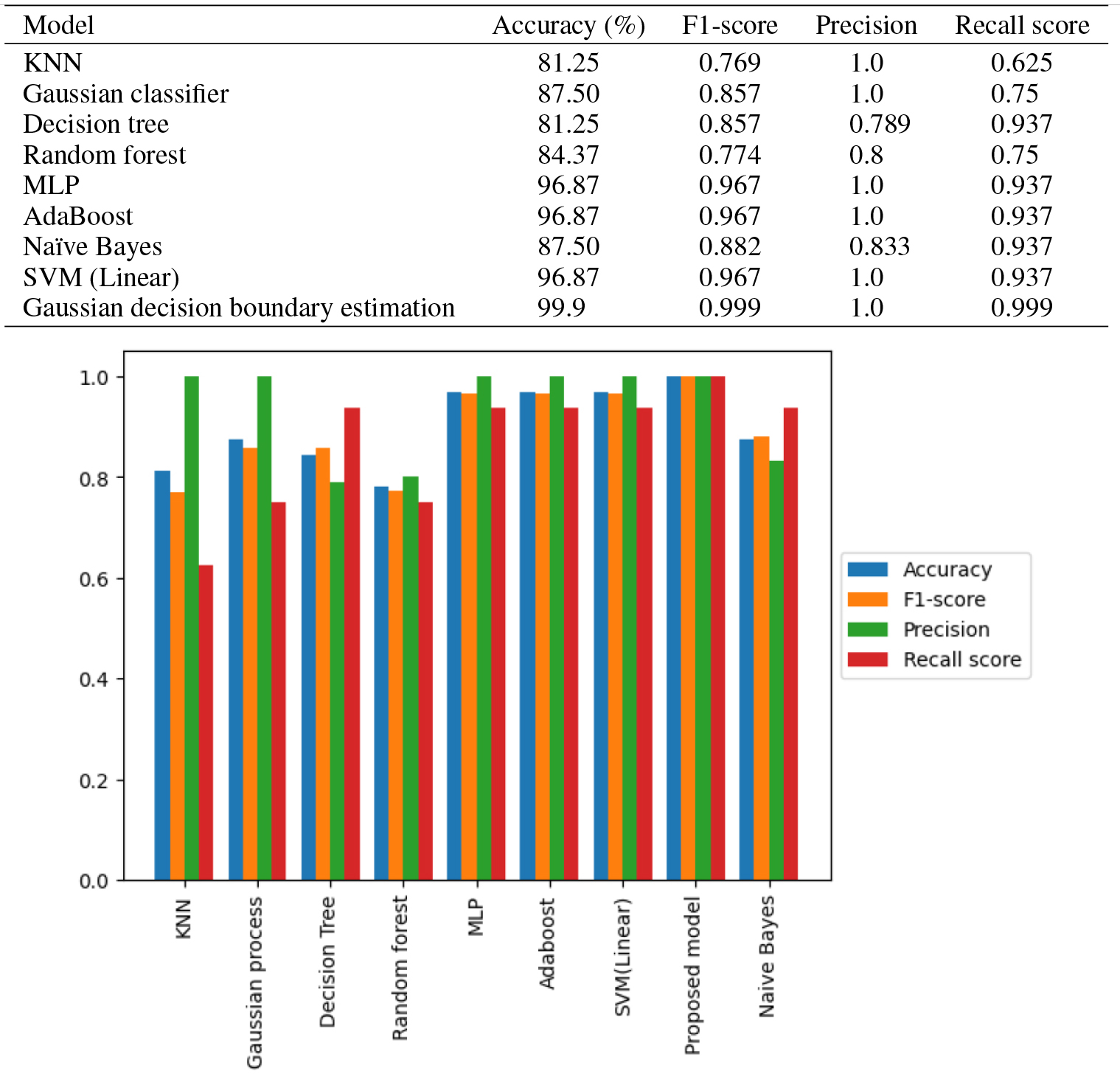
Accuracy, a widely adopted metric in classification performance assessment, delineates the ratio of correctly classified instances to the overall number of instances under consideration. The
Mathematically, the accuracy (Acc) can be defined as:
Where,
TP – number of true positives (correctly predicted positive instances).
TN – number of true negatives (correctly predicted negative instances).
FP – number of false positives (incorrectly predicted positive instances, also known as Type I errors).
FN – number of false negatives (incorrectly predicted negative instances, also known as Type II errors).
The F1 score serves as another widely adopted performance metric for evaluating the efficacy of machine learning models, particularly in the context of binary classification tasks. It acts as a harmonizing factor, striking a balance between precision and recall, thereby furnishing a singular measurement that encompasses both aspects.
Mathematically, the F1 score can be defined as:
The F1 score exhibits a numerical range from 0 to 1, wherein 1 represents the optimal score, denoting flawless precision and recall. A heightened F1 score indicates a more equitable equilibrium between precision and recall, thereby suggesting superior overall model performance. Moreover, the F1 score imposes a penalty on models that disproportionately prioritize precision or recall, facilitating a more equitable assessment of the model’s capacity to render accurate positive predictions while minimizing the omission of actual positive instances.
The Receiver Operating Characteristic (ROC) curve and its associated metric, the Area Under the Curve (AUC), assume pivotal significance in the appraisal of machine learning modules, particularly those tasked with binary classification. The ROC curve, portrayed graphically, elucidates the relationship between true positive rate (TPR) and false positive rate (FPR) at distinct classification thresholds, facilitating a nuanced understanding of the sensitivity-specificity trade-off. Subsequently, the AUC, a scalar value derived from the ROC curve, quantifies the model’s overall discriminatory prowess.
An AUC value approaching unity signifies exemplary performance, while an AUC of 0.5 conveys chance-level prediction. The amalgamation of the ROC curve and AUC confers a comprehensive evaluation of the classification model’s proficiency in discerning between positive and negative instances, rendering them indispensable tools for model selection, comparative analysis, and informed decision-making across diverse machine learning applications. The Confusion matrix and Receiver operating characteristic curve of various models are shown in Fig. 10.
The Area Under the Receiver Operating Characteristic (ROC) Curve (AUC) for the proposed model signifies its outstanding discriminative capability, displaying exceptional performance in effectively distinguishing between the promoter and non-promoter classes. The ROC curve analysis yields insights into the model’s high discriminatory ability, minimal misclassification, excellent predictive power, strong class separation, and reliable ranking of predictions. Figure 11 shows the comparative analysis of the proposed algorithm with other state of art algorithms.
The confusion matrix is a fundamental tool for assessing the performance of a classification model. It provides insights into the model’s strengths and weaknesses, helping practitioners make informed decisions about model selection and optimization. It’s particularly useful in situations where the consequences of false positives and false negatives are different. The confusion matrix provides a comprehensive and quantifiable assessment of how well the model is performing, and offers insights into the model’s strengths and weaknesses, helping to identify where it excels and where it may need improvement.
The ROC curve of MLP and AdaBoost classifiers infers that it is performing very well. It’s characterized by a steep rise in the TPR (True Positive Rate) while maintaining a low FPR (False Positive Rate) for most classification thresholds. The step in the top left corner indicates near-perfect performance for a range of thresholds. The Area Under Curve is close to the maximum value, suggesting that these classifiers have a high probability of correctly ranking positive instances higher than negative ones, which is a good sign.
The ROC curve of the proposed model, shows a very positive sign of the model’s extremely performance. Additionally, an AUC (Area Under the Curve) value of 0.999 indicates exceptionally high discrimination ability. The shape of curve indicates that the model can effectively distinguish between positive and negative instances with minimal misclassification. This is a strong indication of a well-trained and well-performing classification model.
The current realm of research is prominently focused on the utilization of diverse data mining technologies encompassing data mining architecture, the development of ML algorithms, and the exploration of novel data mining analysis functions, tailored for biological information processing. Within this context, an extensive comparison of multiple methods for promoter detection in nucleotide sequences has been undertaken. The unequivocal findings underscore the considerable advantages of the proposed approach, exhibiting a remarkable accuracy of 99.9% on the identical dataset, surpassing the efficacy of existing methodologies. In the fields of bioinformatics and genetics, the classification of promoters from DNA sequences has a variety of applications, including understanding gene regulation, disease research, precision medicine, and bioinformatics research.