
Abstract
Protein–DNA interactions are involved in different cancer pathways. In particular, the DNA-binding domains of proteins can determine where and how gene regulatory regions are bound in different cell lines at different stages. Therefore, it is essential to develop a method to predict and locate the core residues on cancer-related DNA-binding domains. In this study, we propose a computational method to predict and locate core residues on DNA-binding domains. In particular, we have selected the cancer-related DNA-binding domains for in-depth studies, namely, winged Helix Turn Helix family, homeodomain family, and basic Helix-Loop-Helix family. The results demonstrate that the proposed method can predict the core residues involved in protein–DNA interactions, as verified by the existing structural data. Given its good performance, various aspects of the method are discussed and explored: for instance, different uses of prediction algorithm, different protein domains, and hotspot threshold setting.
Keywords
Introduction
The protein–DNA binding interactions are essential activities in gene transcription. In recent years, it has been recognized that gene transcription plays a more significant role than previously thought in the context of protein level control. 1 Thus, there is increasing interest in deciphering the protein–DNA binding interactions, which can be determined by the bacterial one-hybrid system in the past.
With the advent of next-generation sequencing and other modern biotechnology, the protein–DNA binding interaction studies have been accelerated from single DNA-binding protein study to the proteome-wide level; for instance, Weirauch et al have applied protein-binding microarray to comprehensively determine the eukaryotic transcription factor DNA-binding specificity in sequence level. 2 Wong et al have applied multiple expectation maximization for Motif Elicitation 3 to generate the coupling DNA motifs on chromatin interactions in human being. 4 Jolma et al have also applied systematic evolution of ligands by exponential enrichment and chromatin immunoprecipitation sequencing to characterize the DNA-binding specificities of human transcription factors, resulting in 239 distinct binding profiles. 5 The sequence and chromatin determinants surrounding protein–DNA binding interactions have also been studied in the context of transcription factors across different cell lines comprehensively. 6 Tremendous data have been accumulated with the potential for deciphering protein–DNA binding interactions further.
Therefore, it is important to harness and leverage the existing big data to shed proteome-wide lights on the protein–DNA-binding studies; for instance, Wong et al have proposed a computational framework to learn and predict the specificity-determining residue–nucleotide interactions across different DNA-binding families. 7 Pelossof et al have also proposed an approach for learning the recognition models across different DNA-binding families. 8 Wong et al have proposed an evolutionary computational approach for learning the combinatorial protein–DNA-binding sequence patterns. 9
Objective
In this study, we propose to adopt SNPdryad 10 for the prediction of DNA-binding residues. In other words, given a protein sequence, we would like to predict and locate which residues are DNA binding, as verified by its protein–DNA complex structural information from Protein Data Bank (PDB).
Methodology
To predict the DNA-binding residues for a specific DNA-binding domain family, we are interested in the positional distribution of the harmful nonsynonymous single-nucleotide polymorphisms (nsSNPs) across the DNA-binding domain family because nsSNPs are known to be able to directly alter the encoded protein functions. Therefore, we have performed multiple sequence alignment on the protein-coding sequences for each domain and count the number of harmful nsSNPs on these sequences.
The algorithm and the prediction results can be accessed from the website. a A snapshot of the website is shown in Figure 1. In particular, we would like to note that the precise number of dele terious amino acid substitutions on the human proteome is yet to be determined. Thus, the website is designed to remind the users about it when they query the SNPdryad website as shown in Figure A1.

Snapshot of the SNPdryad website in June 2014. The users can download the entire predictions of the human proteomes hg19 and hg18.
In particular, we would like to note that SNPdryad is trained on the existing literature annotations (Harvard HumDiv dataset) that may be limited in size if we consider all the possible amino acid substitutions on a human proteome, although this is also the approach the existing methods (eg, Harvard PolyPhen2) have taken.
Results
Fully Deleterious Substitutions
In total, we scanned 92,012 human proteins (including protein isoforms) and 36,935,804 amino acid positions; a total of 10,120,155 substitutions (about 1.4%) were predicted to be fully deleterious (with the SNPdryad prediction score of 1).
DNA-Binding Family Choices
In this study, we have selected three DNA-binding protein families for in-depth studies. (1) The ETS domain has been selected (Pfam ID: PF00178) since it is important in different tissue developments and cancer progression for metazoans. 11 (2) The homeodomain family is selected because it was demonstrated to be related to multiple cancers: breast cancer, 12 prostate cancer, 13 and nonmuscle invasive bladder cancers. 14 (3) The basic Helix-Loop-Helix (bHLH) domain family is selected because its proteins are found to sense and respond to environmental stimulus in different cancer-related pathways. 15
Winged Helix Turn Helix family
The ETS domain has been selected (Pfam ID: PF00178) since it is a large transcription factor family, which is important in different tissue developments and cancer progression for metazoans. 11 The ETS domain belongs to the winged Helix Turn Helix domain family. It is a DNA-binding domain that has three alpha helices and four beta strands (eg, PF03444). Especially, the domain is characterized by the alternative intervention between the helices and strands.
In particular, we extracted the amino acid sequences annotated as the ETS domains in human proteome. The positions of the harmful nsSNPs were mapped onto each sequence. To provide biological insights, we also downloaded the crystal structure of mouse Elf3 C-terminal DNA- binding domain in complex with type II TGF-beta receptor promoter DNA (PDB ID: 3 JTG).
16
The amino acid chain in that structure was aligned with the ETS domain sequences using MUSCLE with the default parameter setting.
17
The resultant multiple sequence alignment and the mapped number of harmful nsSNPs are depicted in Figure 3. Interestingly, it can be observed that the position distribution of the harmful nsSNPs is not uniform across the domain. To distinguish harmful nsSNP hot spots (tall peaks) from the rest, the mean and standard deviation of the number of harmful nsSNPs are calculated. A position is called a harmful nsSNP hot spot when the number of harmful nsSNPs exceeds the mean plus three standard deviations (ie,


Multiple sequence alignment for the ETS-domain (Pfam ID: PF00178) sequences in human and the amino acid sequence extracted from the ETS-domain structure (PDB ID: 3 JTG Chain A), colored in the Clustalx color scheme.
Homeodomain Family
The homeodomain family is a DNA-binding domain that has three alpha helices connected by short loop structures (eg, PF00046). The defining feature is that one of the alpha helix is found nearly perpendicular to the plane formed by the other two alpha helices. The HOX genes (which belong to the homeodomain family) have been demonstrated to be related to different human cancers 18 ; for instance, the induction of HOXA5 can cause around 300 genes to be unregulated in breast cancer cell lines 12 ; the overexpression of HOXC8 was found to be related to the loss of differentiation in prostate cancer cells 13 ; HOXA9 was demonstrated to be an independent indicator of prognosis in nonmuscle invasive bladder cancers. 14
As an illustrative example, we have selected the structure of Aristaless homeodomain (PDB ID: 3 LNQ) as shown in Figure 4. Similar to the previous section, we have exhaustively extracted all homeodomain sequences in human being and performed the multiple sequence alignment. Once aligned, we count how many harmful nsSNPs can be found at each alignment position as shown in Figure 5. To distinguish harmful nsSNP hot spots (tall peaks) from the rest, the mean and standard deviation of the number of harmful nsSNPs are calculated. A position is called a harmful nsSNP hot spot when the number of harmful nsSNPs exceeds the mean plus three standard deviations (ie,
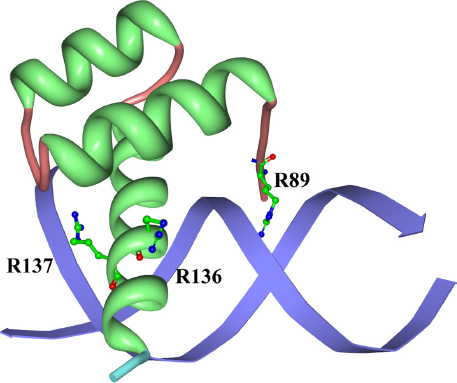
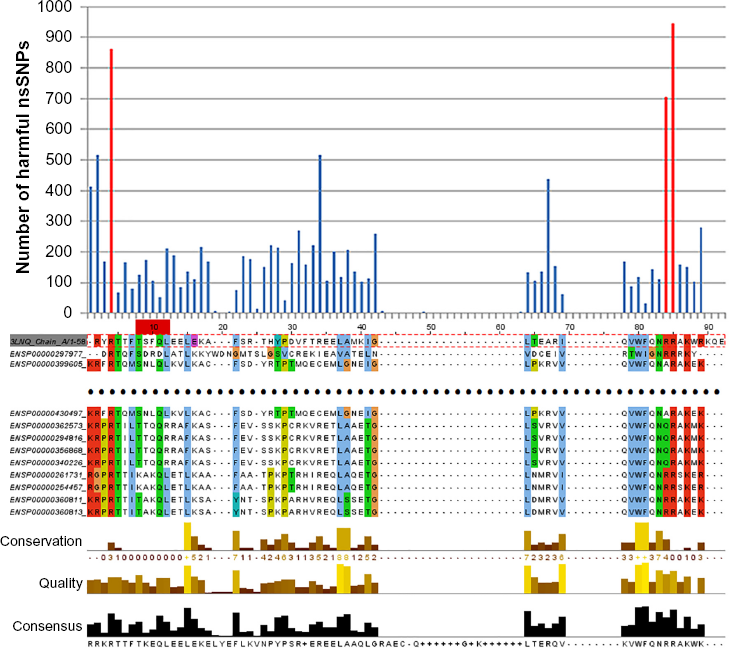
Multiple sequence alignment for the homeodomain domain (Pfam ID: PF00048) sequences in human and the amino acid sequence extracted from the homeodomain structure (PDB ID: 3 LNQ Chain A), colored in the Clustalx color scheme.
bHLH Family
The bHLH domain family is a DNA-binding domain that consists of two alpha helices connected by a loop structure (eg, PF00010). In most cases, they bind to DNA as a dimer (ie, two domains). In addition, they are shown to be related to different cancers; for instance, the transcription factors c-Myc and HIF-1 can interact with each other and promote metabolic advantages to tumor cells 19 and adjust adaptive responses to hypoxic environments. 20 bHLH-PAS proteins are also believed as multitasking family of transcription factors that can sense and respond to environmental stimulus in the cancer-related pathways. 15
Therefore, it is very important to identify the core DNA-binding residues for the bHLH family. In particular, we have selected the crystal structure of MyoD bHLH domain as an example visualized in Figure 6. Similar to the previous sections, we have collected and aligned all the bHLH sequences from the human proteome, resulting in the multiple sequence alignment profile as shown in Figure 7. It can be observed that only one alignment position remains after setting the


Multiple sequence alignment for the bHLH domain (Pfam ID: Pf00010) sequences in human and the amino acid sequence extracted from the bHLH structure (PDB ID: 1MDY Chain A), colored in the Clustalx color scheme.
Discussion
Gene regulation is a very important step in genetics. In particular, gene transcription is responsible for nearly 70% contribution to protein levels. 1 Therefore, it is very important to decipher the gene transcription mechanism in which the protein–DNA-binding mechanism plays a significant role.
To this end, we have proposed a novel approach to predict the core DNA-binding residues on the cancer-related proteins. We propose that SNPdryad can be integrated and transformed to predict and locate core DNA-binding residues for cancer-related protein studies. The results suggest that the approach is feasible and can be explored further.
In the future, we seek to have comprehensive benchmarking on the prediction performance than the existing case studies, although the additional computational burden has to be provisioned carefully. Another interesting direction is to see if other prediction algorithms such as PolyPhen2 and MutationTaster can be applied in a similar fashion. It would be helpful if the existing methods can complement to each other, contributing to accurate ensemble prediction of DNA-binding residues on cancer-related proteins. The peak threshold setting is another interesting direction to be explored for the proposed method in the future.
Author Contributions
Conceived and designed the experiments: KW. Analyzed the data: KW. Wrote the first draft of the manuscript: KW. Developed the structure and arguments for the paper: KW. Made critical revisions: KW. The author reviewed and approved of the final manuscript.
Footnotes
Acknowledgments
The author thanks the four anonymous reviewers for their constructive comments. The author also thanks Tak-Ming Chan for his works in surveying on protein–DNA binding. Last but not the least, the author thanks Zhaolei Zhang for his insightful comments.