
Abstract
Endothelial dysfunction, the earliest manifestation of atherosclerosis, can be initiated by both biochemicals and biomechanical forces. Atherosclerosis occurs predominantly at arterial branch points, arterial bifurcations and the curved segments of great arteries. These are the regions that blood flows turbulently. Turbulence promotes endothelial dysfunction by reducing shear stress upon endothelial cells. The endothelial glycocalyx mediates the effect of shear stress upon the endothelium.
A mathematical analysis of cardiovascular hemodynamics demonstrates that fluid retention increases turbulence of blood flow. While there is no empirical data confirming this relationship, fluid retention is associated with adverse cardiovascular events. Every medical condition that causes fluid retention is associated with increased risk of both atherosclerotic cardiovascular disease and venous thromboembolic disease. In addition, most medications that cause fluid retention are associated with increased adverse cardiovascular effects. Calcium channel blockers (CCBs) and pioglitazone are exceptions to this generalization. Even though data regarding CCBs and pioglitazone contradict the hypothesis that fluid retention is a cardiovascular risk factor, these medications have favorable cardiovascular properties which may outweigh the negative effect of fluid retention.
Determining whether or not fluid retention is a cardiovascular risk factor would require empirical data demonstrating a relationship between fluid retention and turbulence of blood flow. While this issue should be relevant to cardiovascular researchers, clinicians and patients, it is especially pertinent to the pharmaceutical industry. Four-dimensional magnetic resonance imaging and vector flow Doppler ultrasound have the capability to quantify turbulence of blood flow. These technologies could be utilized to settle the matter.
Keywords
Introduction
Endothelial dysfunction is the earliest detectable manifestation of atherosclerosis [1, 2]. Atherosclerosis and venous thrombosis are associated with one another. Consequently, atherosclerosis may induce venous thrombosis or the two conditions may share common risk factors [3, 4]. Proinflammatory cytokines, bacterial endotoxins, viruses, advanced glycation end products generated in diabetes and aging, cholesterol, oxidized lipoproteins that accumulate within arterial walls, vascular endothelial growth factor, estrogens, renin, prorenin, angiotensin II and biomechanical forces initiate endothelial dysfunction [5–9]. In addition, glucose and insulin may directly influence endothelial physiology [10]. Endothelial cell surface receptors bind lipids, estrogens, progestins, mineralocorticoids, thrombin, renin and prorenin [11–17]. When biochemicals bind to endothelial cells, a cascade of intracellular events ensues that culminates in endothelial dysfunction. The main limitation of a biochemical explanatory model of endothelial dysfunction is that atherosclerosis occurs primarily at arterial branch points, arterial bifurcations and at curves of great arteries. Biochemistry cannot explain why atherosclerosis is localized, whereas biomechanics can.
Biomechanics and endothelial dysfunction
Blood flows either in a laminar manner or a turbulent manner. Laminar flow occurs in unbranched, tubular arteries, increases arterial wall shear stress and is atheroprotective. Laminar flow up-regulates endothelial genes that exert antithrombotic, antiadhesive, antiproliferative, anti-inflammatory and anti-oxidant effects [5, 18]. Turbulent flow occurs in regions prone to atherosclerosis: arterial branch points, arterial bifurcations and curved segments of major arteries [19]. Turbulence reduces shear stress upon endothelial cells. As a result, there is increased expression of inflammatory genes and increased production of adhesion molecules by endothelial cells, increased adhesion of platelets to endothelial cells and accelerated atherosclerotic plaque formation [5, 20].
Some of the turbulent blood flow that occurs within the heart is physiological. For instance, whirlpools in the left ventricle shape its walls and are vital for the proper movement and closing of the mitral valve. In addition, in the aortic bulbous, the turbulent flow that occurs during the ejection phase of systole helps to close the semilunar valve, thereby facilitating coronary blood flow. Thus, turbulence and whirlpools are not always negative.
At one time, it was thought that altered hemodynamics in areas prone to atherosclerosis disrupt endothelial integrity and facilitate the penetration of lipoproteins into vessel walls [21]. This hypothesis was discounted after it was recognized that the endothelium remains intact during the early formation of atherosclerotic lesions [22–24]. It has also been suggested that turbulent flow accelerates atherosclerosis by permitting lipoproteins to dwell near the arterial wall for longer time intervals [25].
Endothelial glycocalyx, biomechanics and endothelial dysfunction
The endothelial glycocalyx (EG), a thin polysaccharide layer attached to the lumenal surface of vascular endothelial cells, is composed of glycoproteins and proteoglycans that are synthesized by endothelial cells. Some of these glycoproteins and proteoglycans are anchored to the endothelial cell walls, either via transmembrane domains or by covalent links to molecules associated with the outer portion of the cell membrane [26]. Other components of the EG attach to the endothelium indirectly via receptor molecules [26].
The EG provides a physical and electrostatic barrier between the endothelium and the blood elements, thereby reducing the permeability of the endothelium to erythrocytes, leukocytes, platelets, adhesion molecules, bacteria and viruses [27]. The EG also senses and transduces shear forces via connections to endothelial cell walls and to the cytoskeleton of endothelial cells, thereby mediating the effect of shear stress upon the endothelium [28]. Some of the molecular components of the EG extend perpendicularly from the endothelium in “shrub-like” configurations [29, 30]. These projections act as lever arms that amplify torque and convert shear forces into deformations of the endothelial cystoskeleton [29]. Glycocalyx mechanosensation and mechanotransduction results in activation of endothelial nitric oxide synthase (eNOS), increased production of nitric oxide and dilatation of blood vessels [31].
Increased shear stress promotes the synthesis of glycocalyx components, resulting in a thicker glycocalyx, whereas decreased shear stress causes glycocalyx degradation, resulting in thinner glycocalyx dimensions [32–34]. Areas of the vasculature with a thinner glycocalyx are associated with a pro-inflammatory endothelial cell phenotype and are more vulnerable to atherosclerotic plaque formation [34–36].
Fluid retention, turbulence and cardiovascular disease
Fluid retention increases either cardiac output or blood pressure (BP) [37–39]. Cardiac output is the product of stroke volume and heart rate while cardiac work is determined by stroke volume and BP. A mathematical analysis of cardiovascular hemodynamics demonstrates that fluid retention increases cardiac work while simultaneously increasing velocity and turbulence of blood flow [40]. Although there is no empirical data confirming these relationships, fluid retention is associated with increased risk of adverse cardiovascular events. Figure 1 illustrates a proposed pathway linking fluid retention with endothelial dysfunction and cardiovascular disease.
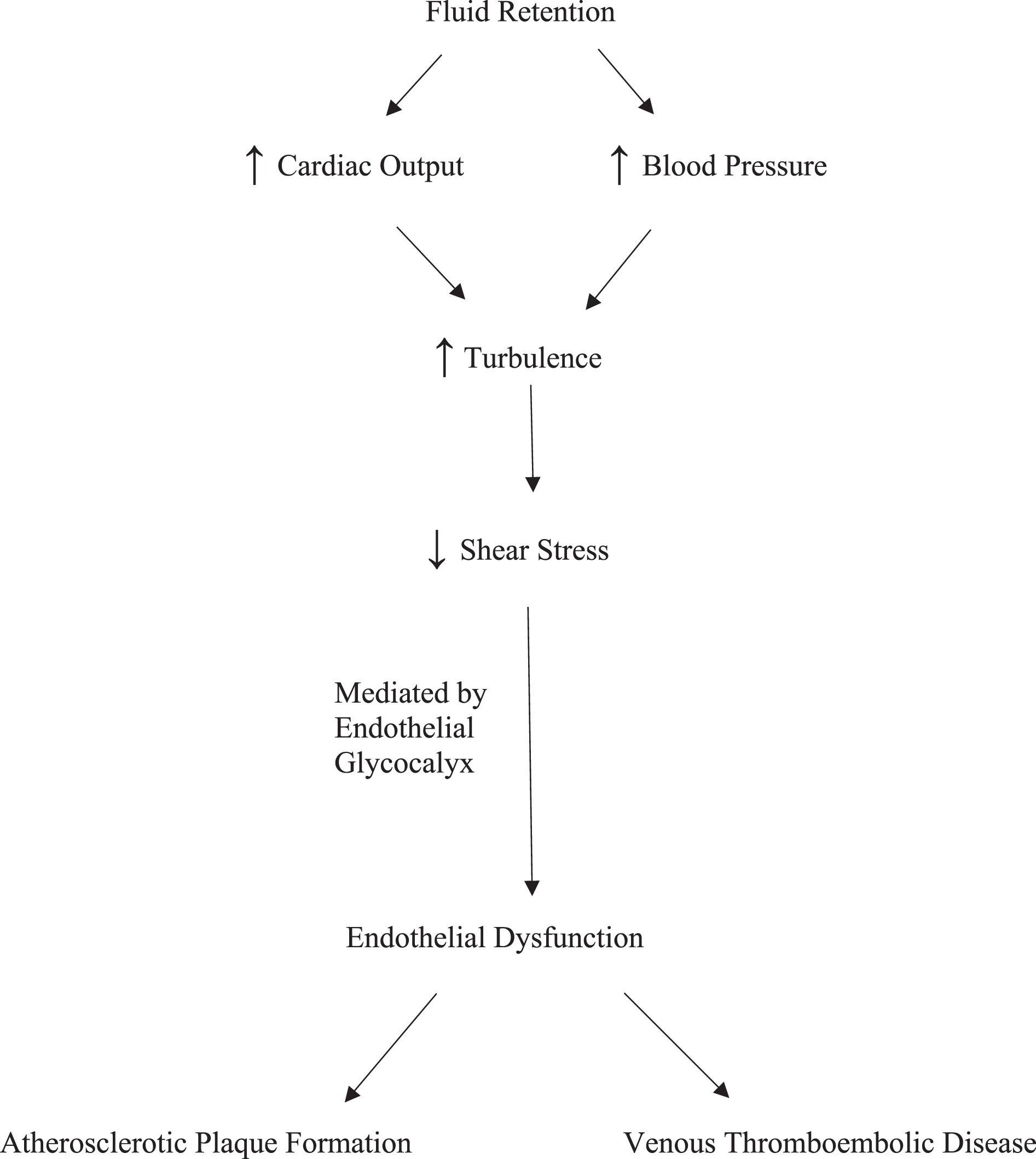
Proposed Pathway Linking Fluid Retention with Endothelial Dysfunction and Cardiovascular Disease.
Every medical condition that causes fluid retention, including heart failure, cirrhosis, nephrotic syndrome, hypothyroidism, type 2 diabetes mellitus (DM), prediabetes, obstructive sleep apnea, hyperaldosteronism and Cushing’s syndrome, is associated with increased risk of atherosclerotic cardiovascular disease (Table 1) [41–56]. With the possible exception of type 2 DM, all these medical conditions are also associated with venous thrombotic events (VTE) [41–56]. Two meta-analyses found a relationship between type 2 DM and VTE but a more recent meta-analysis did not [57–59]. In addition, a Mendelian randomization analysis found no relationship between type 2 DM and VTE [60].
Cardiovascular disease and medical conditions that cause fluid retention
The medical literature is equivocal as to whether or not sex-specific differences influence the cardiovascular complications of fluid retention. For example, one study found that men with prediabetes experience a higher frequency of cardiovascular comorbidities than women with prediabetes while another study found that cardiovascular risk was similar for men and women with prediabetes [61, 62].
For some medical conditions that promote fluid retention, particularly Cushing’s syndrome, type 2 DM and prediabetes, biochemistry is relevant. Mineralocorticoids have direct effects upon endothelial cells [15]. Since corticosteroids have mineralocorticoid activity, the increased corticosteroids in Cushing’s syndrome may have direct endothelial effects. With type 2 DM and prediabetes, glucose and insulin may have direct effects upon endothelial physiology [10]. With hypothyroidism and aldosteronism, it is not likely that biochemistry directly influences endothelial function. There are so few thyroid hormone receptors on the surface of endothelial cells that they are unlikely to be of functional significance [63]. Similarly, aldosterone does not alter the gene expression of endothelial cells [64].
After renin and prorenin bind to endothelial cell surface receptors, endothelial genomic activity increases production of proinflammatory and profibrotic proteins [17, 65]. Fluid retention decreases renin release by the juxtaglomerular apparatus of the kidneys. To the extent that renin and prorenin binding to endothelial cells influences endothelial function, there should be a reduction of inflammatory and fibrotic protein expression by endothelial cells in response to fluid retention. Since fluid retention is associated with increased thromboembolic cardiovascular disease, renin and prorenin probably do not have much direct effect upon endothelial function following fluid retention.
One obvious cause of fluid retention is sodium chloride (salt) consumption. Higher salt intake is associated with increased risk of cardiovascular disease [66, 67]. This relationship is usually attributed to the hypertensive effect of salt ingestion. Nonetheless, increased intravascular volume without hypertension, a phenomenon that occurs in salt resistant individuals, might also be a contributory factor [39, 40]. Brain natriurtic peptide (BNP) and N-amino terminal fragment of prohormone BNP (NT-proBNP) levels reflect intravascular volume and BP. For this reason, BNP and NT-proBNP are used clinically to determine the incidence and severity of heart failure. In the absence of heart failure, and after adjusting for hypertension, higher BNP levels and higher NT-proBNP levels are associated with greater risk of cardiovascular disease [69, 70].
A number of medications cause fluid retention, including cyclo-oxygenase-2 inhibitors (COX-2 inhibitors), non-selective non-steroidal anti-inflammatory drugs (NSAIDs), corticosteroids, hormone replacement therapy with estrogens and progestins, oral contraceptives, insulins, sulfonylureas, gabapentin and pregabalin. As shown in Table 2, all these medications are associated with increased risk of thromboembolic cardiovascular disease [71–93]. Insulins, corticosteroids, estrogens and progestins may have direct effects upon endothelial cells [10, 12–15]. Rofecoxib increases cardiovascular risk even though it decreases some markers of inflammation associated with endothelial dysfunction [94]. This apparent contradiction is reconcilable by the observation that the cardiovascular risk of rofecoxib correlates with elevations in pro-BNP [95].
Cardiovascular disease and medications that cause fluid retention
Cardiovascular disease and medications that cause fluid retention
Androgen therapy promotes fluid retention but the research literature is mixed concerning cardiovascular risk. Retrospective studies have documented that testosterone supplementation is associated with a reduction of cardiovascular risk while others have documented the opposite [96, 97]. A prospective study found that testosterone replacement therapy has no effect upon atherosclerotic cardiovascular events but increases the incidence of pulmonary emboli [98].
Several categories of antihypertensive medications cause fluid retention but improve cardiovascular outcomes. For calcium channel blockers (CCBs), alpha receptor antagonists and hydralazine, cardiovascular outcomes are more dependent upon BP reduction than fluid retention. Even so, if fluid retention contributes significantly to adverse cardiovascular outcomes, one would expect better cardiovascular outcomes with diuretics than with CCBs. This is not the case. Accordingly, either the hypothesis that fluid retention contributes to adverse cardiovascular events is flawed or else other factors play a more important role.
Antihypertensive medications have different effects upon lipids and BP variability. It is possible that these properties outweigh the effects of fluid retention. Diuretics raise cholesterol, angiotensin converting enzyme (ACE) inhibitors have a neutral effect on cholesterol in non-diabetic patients, and CCBs have a neutral effect on cholesterol [99]. Increased BP variability is associated with adverse cardiovascular outcomes [100]. Amlodipine reduces BP variability more than other CCBs, and CCBs reduce BP variability more than other classes of antihypertensive medications [101–103]. Non-loop diuretics reduce BP variability while ACE inhibitors, angiotensin receptor blockers (ARBs) and beta-blockers increase BP variability [101–103].
In the CAMELOT trial, the frequency of adverse cardiovascular outcomes was lower in the amlodipine cohort than the enalapril cohort [104]. The combination of benazepril and amlodipine in the ACCOMPLISH trial was more effective than the combination of benazepril and hydrochlorthiazide (HCTZ) in reducing adverse cardiovascular events despite slightly greater BP lowering in the benazepril-HCTZ group [105]. In addition, INSIGHT demonstrated equivalence in cardiovascular outcomes comparing nifedipine versus a diuretic while NORDIL found that diltiazem is more effective than diuretics or beta-blockers at preventing strokes [106, 107].
Not all clinical trials are consistent with a hierarchy of antihypertensive medications based upon BP variability and lipid properties. ACE inhibitors are more effective than non-amlodipine CCBs at preventing coronary heart disease, heart failure and major cardiovascular events [108]. Studies have shown that non-amlodipine CCBs are inferior to other types of antihypertensive drugs as first-line agents in reducing the risks of several major complications of hypertension [109]. In the ALLHAT trial, chlorthalidone was found to be comparable or superior to lisinopril and amlodipine in terms of several adverse cardiovascular events but BP reductions were not equivalent among the chlorthalidone, lisinopril and amlodipine cohorts [110].
Thiazolidinediones
Pioglitazone and rosiglitazone both cause fluid retention but have discordant effects upon cardiovascular outcomes (Table 2). Pioglitazone decreases the risk of myocardial infarcts and strokes while rosiglitazone increases the risk of myocardial infarcts [111–113]. Rosiglitazone also increases the risk of cardiovascular death to a degree that borders on significance [113]. The favorable effect of pioglitazone upon cardiovascular outcomes contradicts the hypothesis that fluid retention increases cardiovascular risk. Consequently, either the hypothesis is flawed or else other factors play more important roles.
Pioglitazone and rosiglitazone improve glycemic control and modestly decrease BP [114, 115]. The discordant cardiovascular effects of pioglitazone and rosiglitazone may be a result of their differing effects upon lipids [114]. Pioglitazone has a neutral effect upon low-density lipoprotein (LDL) -cholesterol whereas rosiglitazone raises LDL-cholesterol [114]. Pioglitazone lowers triglyceride levels whereas rosiglitazone has a neutral effect upon triglycerides [114].
It is possible that for both pioglitazone and rosiglitazone, BP lowering and improved glycemic control favorably influence cardiovascular outcomes, fluid retention negatively influences cardiovascular outcomes, and lipid effects are responsible for their divergent cardiovascular effect. For pioglitazone, it may be that the combined effect of its BP, glycemic, fluid and lipid properties decreases adverse cardiovascular outcomes. For rosigltiazone, it may be that the combined effect of these properties increases adverse cardiovascular outcomes.
Implications for pharmaceutical industry and regulators
The possibility that fluid retention contributes to cardiovascular risk may have relevance for the pharmaceutical industry and for regulatory agencies such as the Food and Drug Administration (FDA). Since hypertension and elevated LDL-cholesterol are recognized cardiovascular risk factors, the FDA could require BP data, cholesterol data and fluid retention data for new drug applications. This might allow the FDA to predict the cardiovascular risk of medications. When warranted, cardiovascular safety data could be required prior to regulatory approval or, alternatively, the FDA could mandate an appropriate warning on the drug label.
Empirical evidence of turbulence
The cardiovascular effects of pioglitazone and calcium channel blockers cast doubt upon the hypothesis that fluid retention is a cardiovascular risk factor. Absence of empirical data is an additional limitation. Four-dimensional magnetic resonance imaging (MRI), as well as vector flow Doppler ultrasound, have the capability to quantify turbulence of blood flow [116, 117]. These technologies could be used to clarify the effect of fluid retention. Measuring turbulence of blood flow before and after fluid administration, or measuring turbulence of blood flow before and after prescribing medications that cause fluid retention, would enable researchers to determine the effect of fluid retention.
Conclusion
In summary, biochemicals and biomechanical forces trigger endothelial dysfunction but only biomechanics can explain why atherosclerosis is localized to arterial branch points, arterial bifurcations and the curved segments of great arteries. These are the regions that blood flows turbulently. Turbulence promotes endothelial dysfunction by reducing shear stress upon endothelial cells. The endothelial glycocalyx mediates the effect of shear stress upon the endothelium.
A mathematical analysis of cardiovascular hemodynamics demonstrates that fluid retention increases turbulence of blood flow. However, there is no empirical evidence of this relationship. Medical conditions that increase fluid retention are associated with increased risk of arterial and venous thromboembolic cardiovascular disease. Likewise, many medications that cause fluid retention either increase cardiovascular risk or are associated with increased cardiovascular risk. While the favorable cardiovascular effects of pioglitazone and calcium channel blockers contradict the hypothesis that fluid retention is a cardiovascular risk factor, these medications have properties that may compensate for fluid retention.
The primary limitation of the hypothesis that fluid retention is a cardiovascular risk factor is the absence of empirical data demonstrating that fluid retention increases turbulence of blood flow. Four dimensional MRI and vector flow Doppler ultrasound technologies could be utilized to settle the matter.