
Abstract
BACKGROUND:
Endogenous retroviruses, previously deemed “junk” DNA, have gained attention in recent scientific studies. These inherited genomic elements are now recognized for their potential roles in diseases, especially cancer, highlighting their value as potential diagnostic or therapeutic targets.
OBJECTIVE:
This research aims to explore the association between human endogenous retroviruses (HERV) and gastric cancer, focusing on discerning HERV expression patterns and understanding their implications in gastric cancer pathology.
METHODS:
A quantitative analysis of HERV expression was conducted, employing Support Vector Machine (SVM) and AdaBoost algorithms to identify discriminative HERVs. The co-regulation network between protein-coding genes and HERVs was constructed using the Weighted Gene Co-expression Network Analysis (WGCNA).
RESULTS:
Three distinct HERVs (LTR16A
CONCLUSIONS:
HERVs appear to influence abnormal inflammatory responses, suggesting a pivotal role in gastric cancer development.
Introduction
Human endogenous retroviruses (HERVs) are remnants of ancient retroviral infections and constitute nearly 9% of the human genome [1]. These retroviral elements integrated into the human genome and accumulated mutations, becoming an integral part of the genome. The canonical HERV genome contains three genes (gag, pol, and env) and is flanked by long terminal repeat (LTR) sequences which contain regulatory elements [2]. While most HERVs are usually repressed and considered to be non-functional, they can be triggered by various endogenous and exogenous factors [3].
As a significant health concern, gastric cancer ranked as the sixth most prevalent cancer and the third leading cause of cancer-related fatalities worldwide [4]. In 2020, approximately 1,089,103 new cases were diagnosed, and about 768,793 patients succumbed to the disease [4]. The main recognized causes of gastric cancer include Helicobacter pylori infection [5], various high-risk genetic mutations [6, 7], and environmental factors [8].
Recent studies found that aberrant HERV expression may be associated with human cancers [3]. Despite of their potential significance, endogenous viruses have not been extensively studied due to their integration as a part of the genome and epigenetically silenced under routine conditions [9]. Most studies on the relationship between viruses and tumors have not focused on HERV on human genome. For example, in the study of gastric cancer pathogenesis, much research has focused on the mechanisms of microbial infection, such as Helicobacter pylori or EBV [10], or on the differential expression of protein-coding genes, such as ERBB2 [11] or VEGFR2 [12].
Several studies have investigated the potential association between endogenous retroviruses and cancer development. Yu et al. demonstrated that LTRs of the HERV-W family may cause human uroepithelial cell carcinoma, in which mutant sequences of LTR bind to c-Myb and promoted the expression of Syncytin-1, leading to increased proliferation and viability of human uroepithelial cells [13]. Nakagawa et al. found the expression of ERV3-1 was upregulated in acute myeloid leukemia (AML) patients, suggesting a potential association between HERV and AML [14]. Dolci et al. found that HERV differentially expressed in advanced colon cancer patients and suggested that Env proteins may promote cancer progression [15]. Curty et al. found a significant positive correlation between HERV expression and RAD50 and AC060780.1 in the breast cancer microenvironment, suggesting that HERV may play a role in regulating breast cancer associated genes in HIV carriers [16]. These studies suggest endogenous retroviruses indeed participated in cancer development and worth exploring.
In early studies targeting HERVs as potential bio-markers for tumors, PCR technology was the main method used, focusing on a limited range of HERVs, such as studies on HERV-P, HERV-H, and HERV-K as non-invasive biomarkers for lung cancer [17], and using real-time PCR to study the expression differences of HERV-R, HERV-H, HERV-K, and HERV-P in the blood of breast cancer patients [18]. However, with the gradual reduction in the cost of second-generation sequencing, research on HERVs has entered the realm of large data volumes and multiple types, such as using publicly available datasets of RNA-seq raw data from Alzheimer’s disease patients to perform quantitative counting of all HERVs and compare them with healthy controls to find differences in HERVs. It has been found that the expression of HERVs is spread throughout the genome and may affect the normal expression of genes [19].
Our study provides a novel approach to analyze HERV expression in different tumors and explores their potential impact on gene expression. By examining HERV expression in a larger scale and analyzing their correlation with gene expression, we gained a better understanding of the role of HERV in gastric cancer development and progression. The findings of this study may contribute to the development of novel diagnostic and therapeutic strategies.
Materials and methods
Compilation and analysis of transcriptome datasets
Transcriptome sequencing datasets were compiled from various sources, starting with 56 paired samples from gastric cancer patients, obtained from the NCBI database under the accession number PRJNA435914. To further validate the biomarkers identified, additional data from a separate gastric cancer research project (PRJNA1013242), comprising 61 normal and 48 tumor samples (totaling 109 cases), was also included. For lung cancer, datasets from 20 paired samples were retrieved under the project PRJNA344450 [20], providing a comparative analysis across different cancer types. The liver cancer dataset (PRJNA598835 [21]) included 35 tumor and 30 normal samples, offering insights into the expression patterns in hepatic malignancies. Breast cancer research data (PRJEB50623) contributed to 31 tumor and 10 normal samples, enriching the diversity of the dataset. Pancreatic cancer samples (PRJNA490335 [22]), with an equal distribution of 10 tumor and 10 normal samples, and colorectal cancer data (PRJNA288518 [23]), with 5 tumor and 5 normal samples, were also compiled. These datasets served as an external control to validate the specificity and reliability of the biomarker expression levels identified in our study.
HERVs annotation and differential expression analysis
The various types of HERVs were identified on the human-reference genome (GRch38) using RepeatMasker software (V4.0.9_p2) and Dfam gene component database [24]. To ensure that the HERV position is unique, the combination of the HERV name and position was adopted as keywords in querying the annotation to display.
The differential expression analysis of HEARVs. (a) Differential HERVs in the gastric cancer dataset were screened based on the criteria of 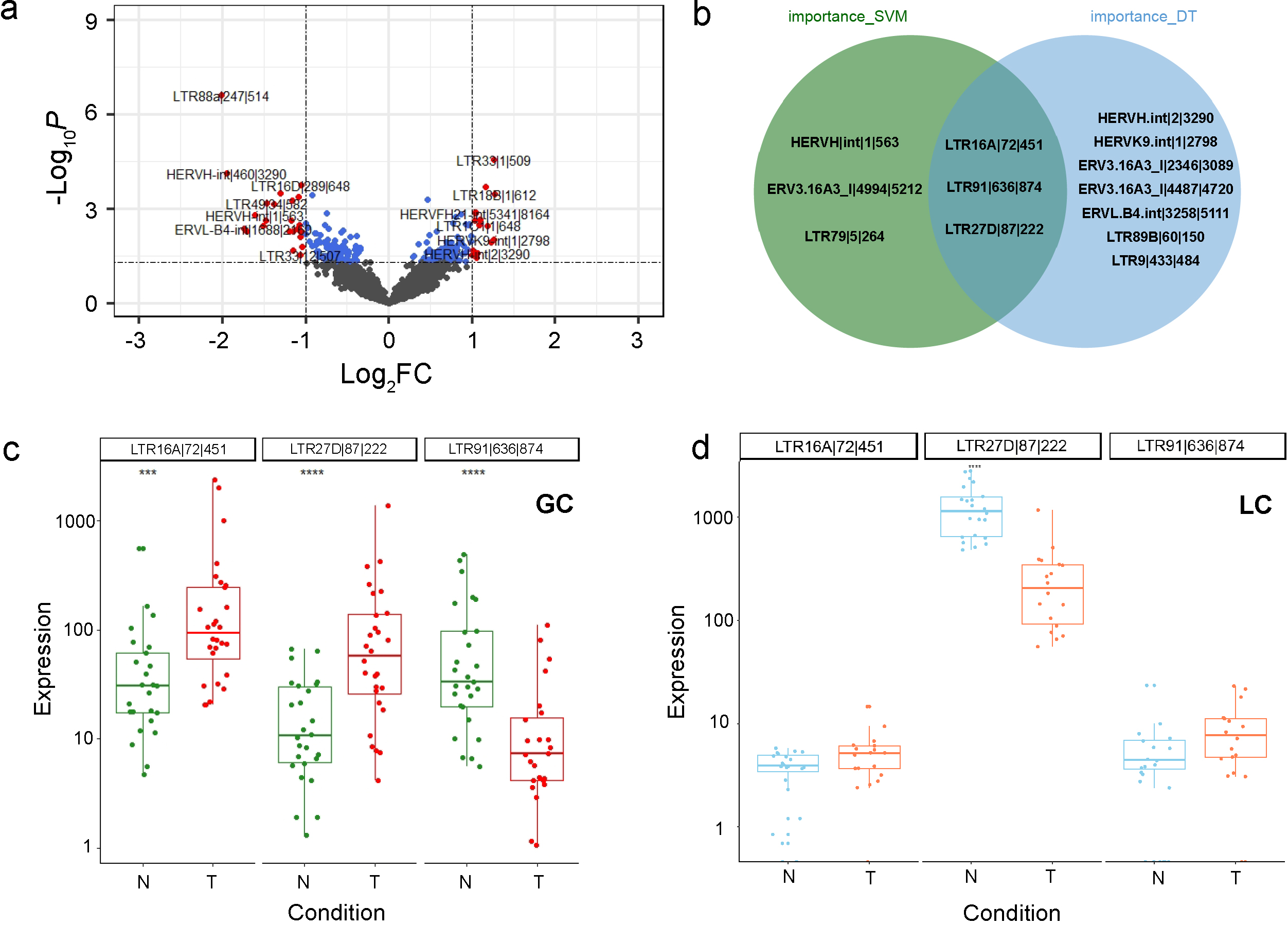
In the transcriptome analysis, the HERVs sequences were retrieved and decompressed to fastq, and the Hisat2 (V2.2.1) [25] was adopted to query the HERVs data against the reference genome(GRch38). The Featurecounts (V2.0.1) [26] was used to quantify the aligned reads of each protein-coding and non-coding genes using EM algorithm. Based on the count matrix, the differential expression analysis was performed by DESeq2 package (V1.32.0) [27], and a combination of
In the feature selection process, a combination of SVM [28] and AdaBoost [29] algorithm was employed to develop an efficient, interpretable, and robust statistical model. This method created perturbed samples by randomly altering the sample labels and multivariate screening of variance fractions. The importance of each feature in discriminating cancer group from control groups was then determined and the mean value of each feature was subsequently calculated. By several cycles of eliminating the least significant feature and progressively reducing the features, a combination with the smallest variance was obtained. This approach enhanced model performance during the feature selection process, capitalizing the strong interpretability of linear SVM and generalization performance of AdaBoost.
The WGCNA analysis of protein-coding genes related with HERVs-elevation. (a) the correlation between gene sets identified by WGCNA and LTR16A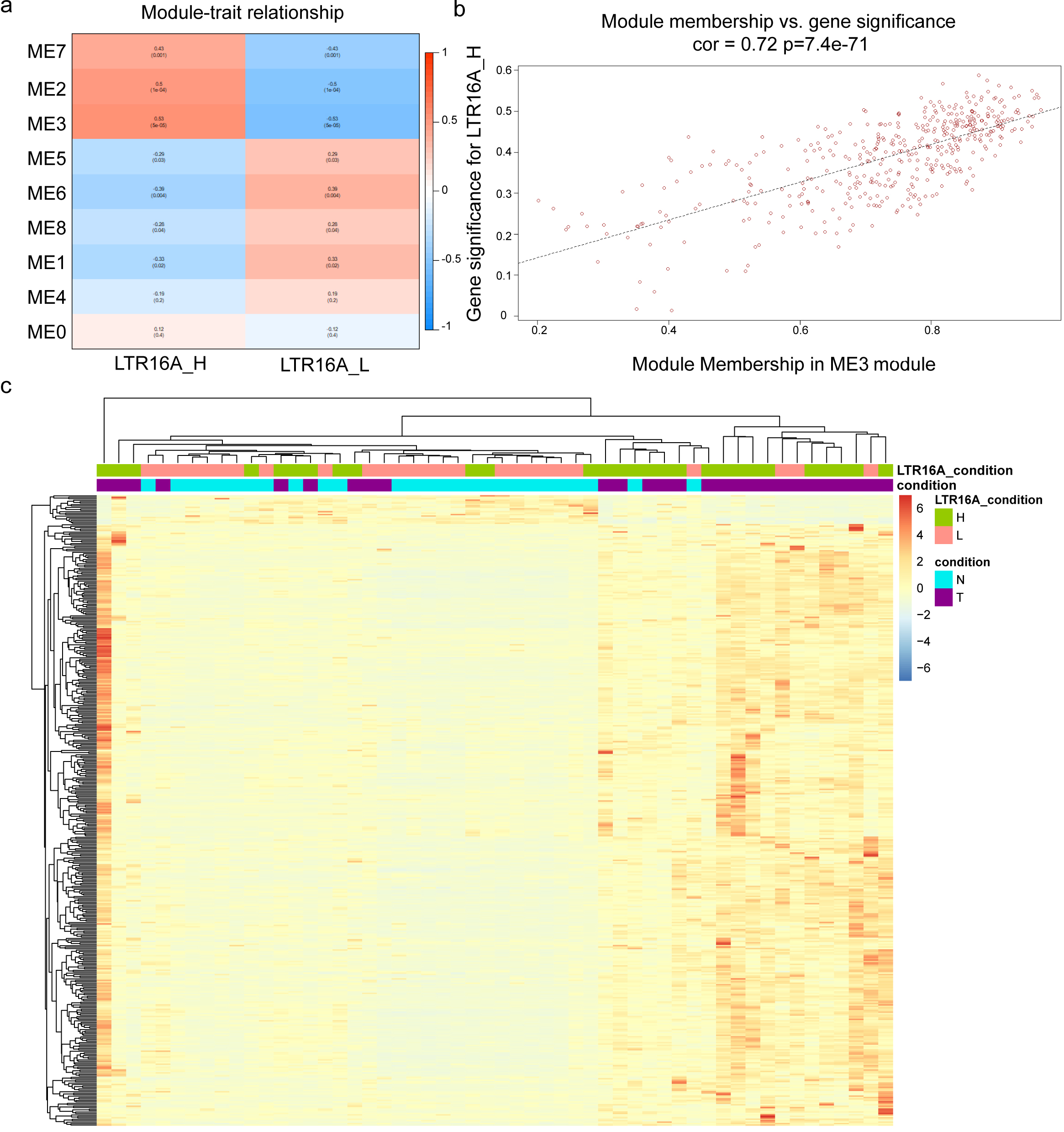
According to the selected HERV expression intensity, the samples were divided into high and low groups, and an correlation analysis between HERV and genes was performed using the WGCNA package (V1.71) [30]. The set of genes with a high correlation with the high and low expression of HERV were compiled and the correlation network was constructed. The pathway processes were analyzed using Clusterprofiler (V4.0.5) [31] to determine the biological function of the gene sets. Finally, we utilized the chooseTopHubInEachModule function from WGCNA to identify the genes with the highest connectivity within the entire gene set. Additionally, we employed gastric cancer data from TCGA to examine the impact of these genes on survival.
Genomic positions and the correlation network of protein-coding genes associated with HERVs-elevation, From the outermost ring, the first ring represents chromosomes, the second ring represents the coordinates of genes, the third ring represents the expression status, and the central position connecting lines represent the weight relationships among genes based on WGCNA results. Red lines represent correlations within the same chromosome, blue represents interrelationships between chromosomes.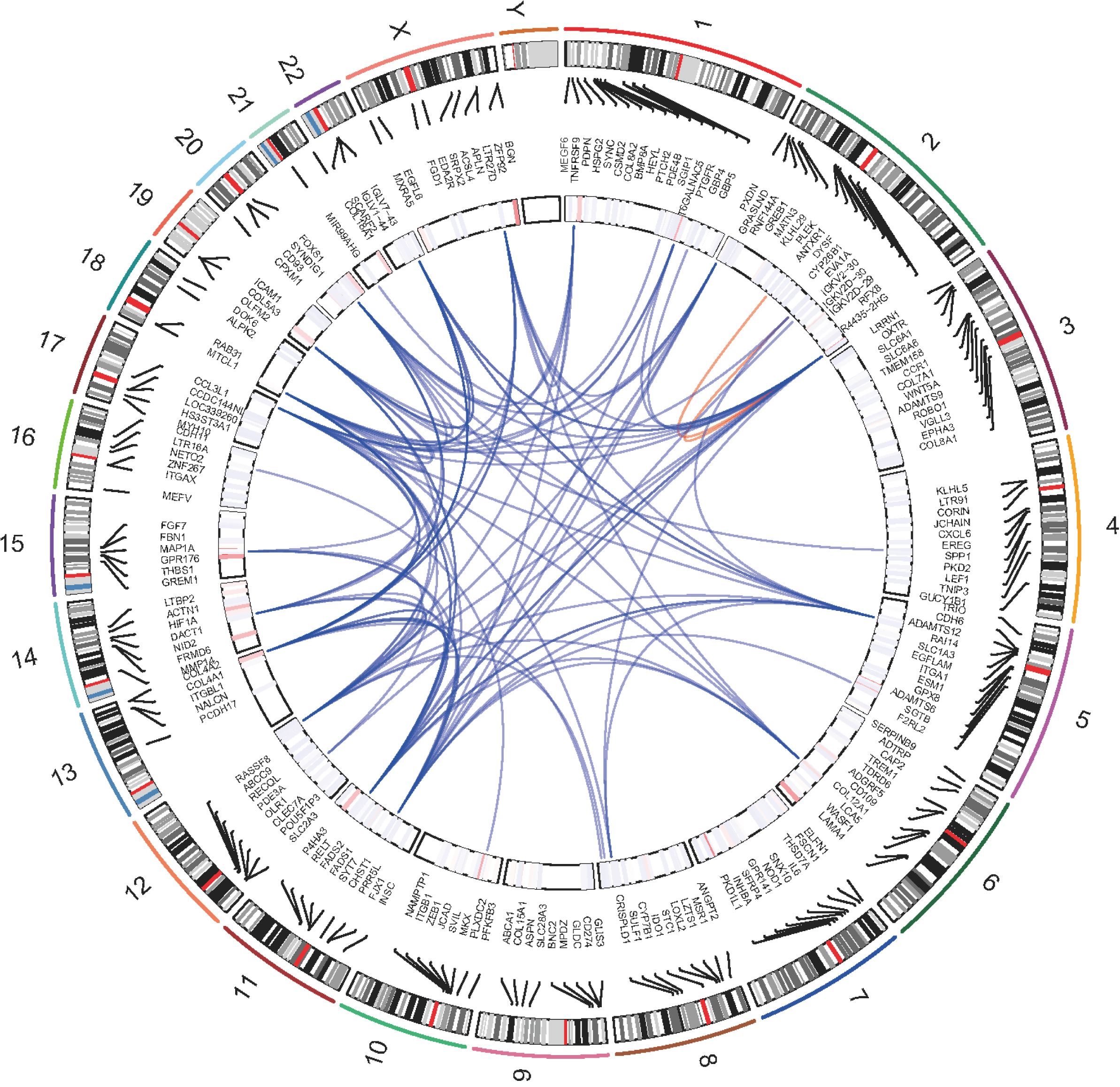
Differential expression of HERVs in gastric cancer and screening of biomarkers
We compiled the transcriptome datasets of gastric cancers and processed the raw expression matrix using DESeq2. The significantly expressed genes were identified between cancer and normal groups (Fig. 1a), in which 22 HERVs were upregulated and 18 HERVs were downregulated.
To identify potential HERV markers for gastric cancer, we carried out a multivariate screening process using the SVM model and AdaBoost algorithm. The LTR16A
Functional analysis of HERV-elevation related genes based on (a) KEGG pathway (b) GO molecular function domain (c) GO biological process domain (d) GO cellular component domain.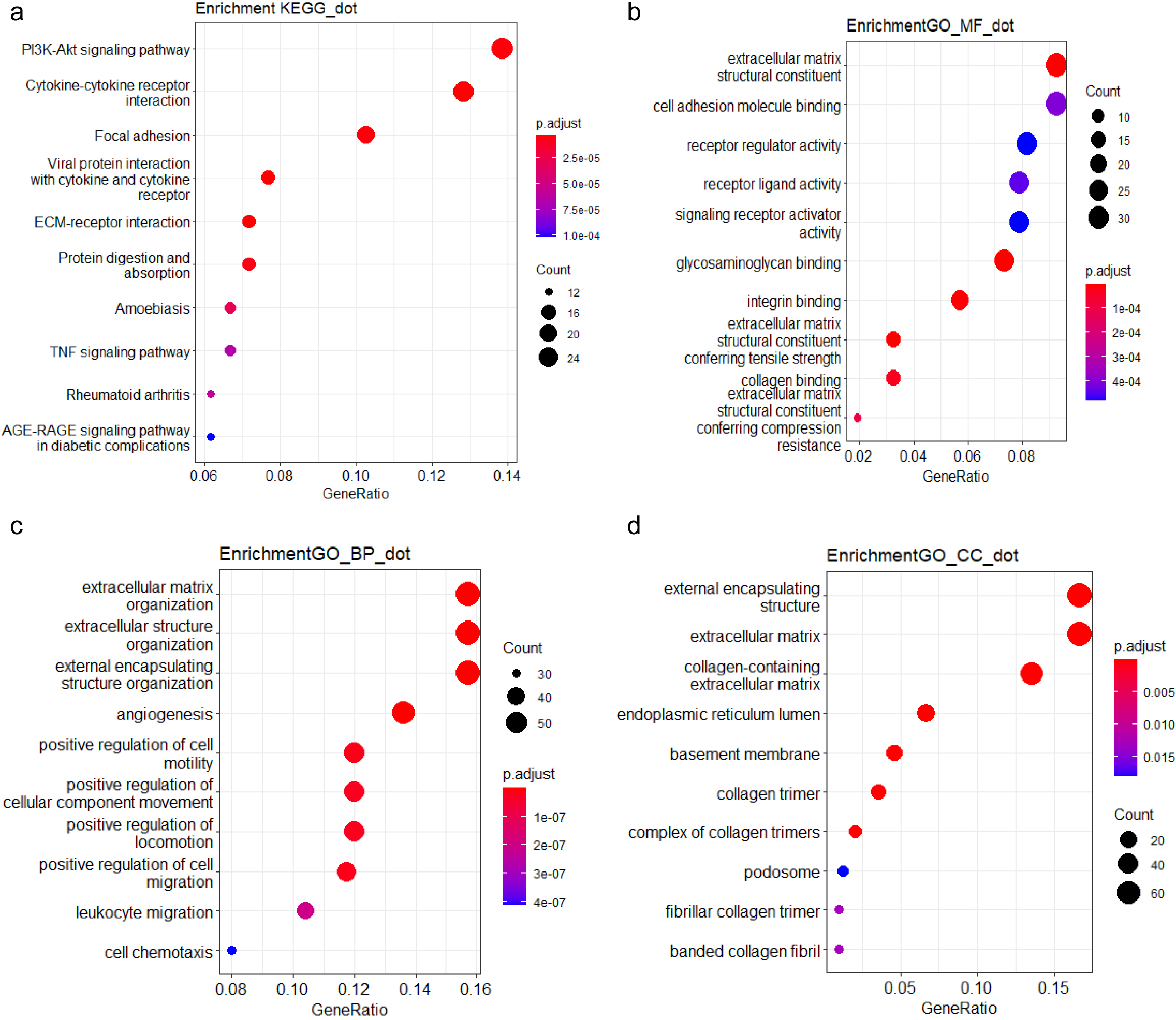
GSEA of HERV-elevation related genes. (a) Profile of the running ES score and the gene set members on the rank ordered list. (b) The functional relationships among the HERV-elevation related pathways.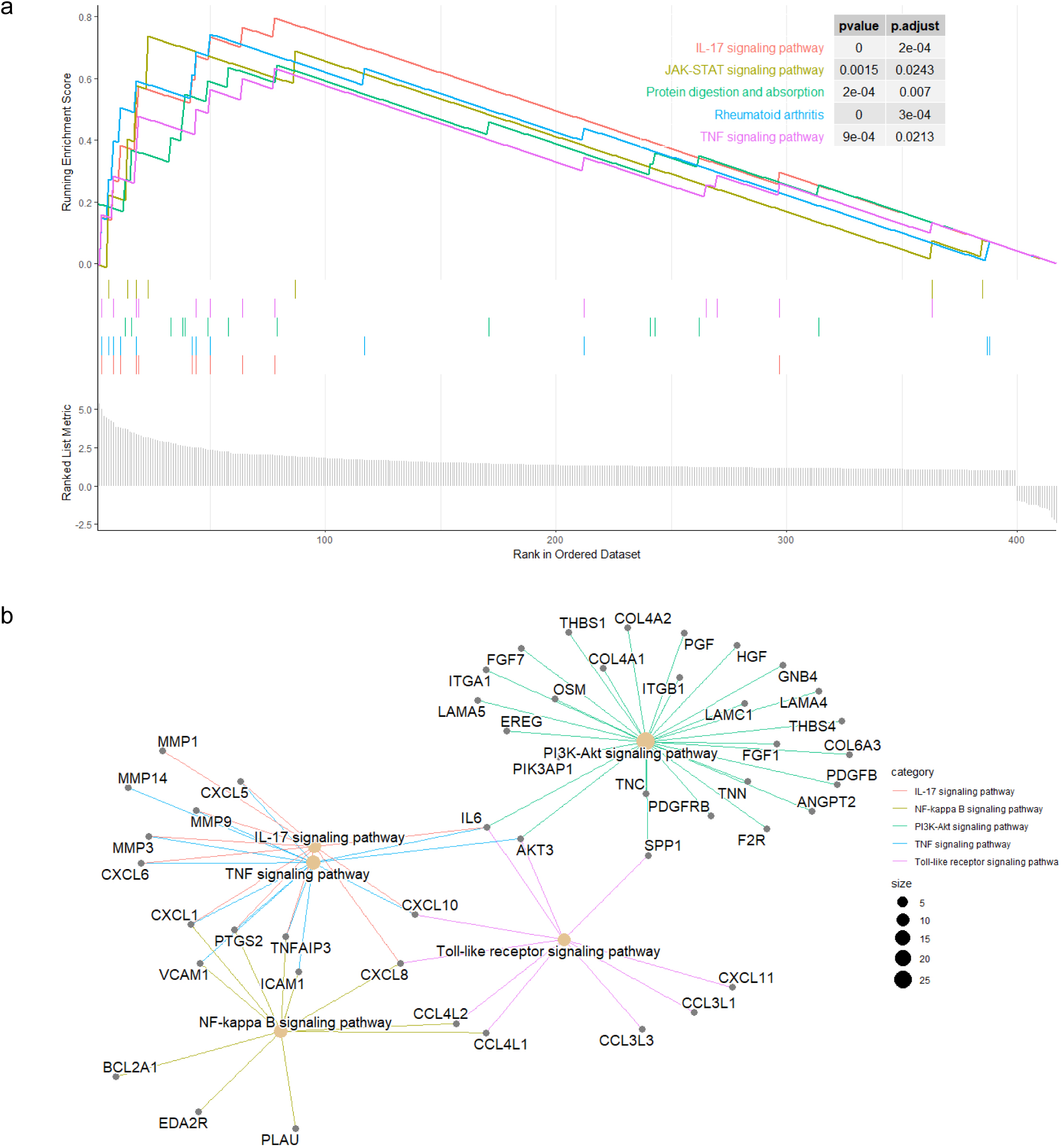
To further investigate the relationship between HERV biomarkers and gene expression, we used the median expression levels of these three biomarkers as a standard for high and low grouping. Combined with the previous expression level box plots, we found that the biomarker LTR16A
Weighted genes co-expression network analysis (WGCNA) analysis was performed to determine the relationship between HERV expression and gene expression in the high and low expression groups, respectively. The Principal Component Analysis (PCA) of the gene expression data within a specific module generated the Module Eigengene (ME) and represented the overall expression characteristics of the genes. In brief, genes with similar expression patterns were grouped into modules, PCA was then performed on the gene expression data for each module. The first principal component captured the largest portion of variation in the gene expression data and was used as the Module Eigengene. The ME carried the overall expression pattern within the module and was utilized to evaluate the correlation between the module and phenotypic traits, in this way, the gene modules significantly associated with traits could be identified.
We displayed the correlation between the gene sets after clustering and traits in Fig. 2a, where a high correlation between the gene set represented by ME3 and the high expression of LTR16A is clearly visible. Subsequently, the correlation between all genes within the ME3 module and the condition of high expression of LTR16A
According to the WGCNA results, we retrieved the interaction relationships between genes within the corresponding gene set (Fig. 3). We selected the weight greater than 0.4 as cutoff and constructed the gene correlation network. The network degrees were computed, identifying RAB31 as a central hub. This designation of RAB31 as the hub gene was achieved using the ‘chooseTopHubInEachModule’ function. This function operates by selecting genes within distinct modules and assessing the correlation among these genes based on their expression data. These correlations are subsequently converted into measures of connectivity strength, which are used to construct an adjacency matrix. Through this process, RAB31 was pinpointed as the gene exhibiting the highest connectivity within its module, establishing its role as a key hub.
Functional analysis of protein-coding genes correlated with HERV expression elevation
To further explore the biological functions and pathways related by the genes with high correlation to HERV elevation, we performed KEGG and GO analysis on this gene set. The KEGG analysis revealed that the genes associated with the three potential HERV markers were mainly enriched in the PI3K-Akt pathway, which is critical to tumorigenesis (Fig. 4). We also performed Gene Set Enrichment Analysis (GSEA) to score the identified pathways using the foldchange information. The top five pathways with significant enrichment showed an overall upregulation trend. Among them, the IL-17 signaling pathway showed the most significantly enriched and followed by the JAK-STAT and TNF pathway (Fig. 5a).
Subsequently, we investigated the interrelationships of several major pathways in Fig. 5b. The genes associated with the gene set were mainly enriched in the IL-17 pathway, and several major pathways belong to typical inflammatory response pathways [32]. We can clearly see the process starting from the IL17 pathway, affecting TNF through genes such as MMP3 and CXCL1, and then extending to NF-kappa B through genes like CXCL1 and VCAM1. Meanwhile, IL17 also connects to the PI3K-Akt pathway through IL6 and AKT3, broadly affecting more related genes. In this process, we can observe several hallmark tumor pathways that influence and interact with each other, closely linked.
Survival analysis of key genes RAB31 which correlated with HERV expression elevation
To investigate the functional impact of HERV-related genes on the survival of gastric cancer patients, we identified RAB31 as the hub gene within our selected gene set, proposing it as a candidate prognostic marker. The gene RAB31 in gastric cancer can also be a potential therapeutic target [33, 34]. We obtained a gastric cancer dataset from TCGA and used the median expression level of RAB31 as the basis for dividing into high and low expression groups. The results of the survival analysis based on these groups were then presented in Fig. 6.
Survival analysis of the RAB31 gene in TCGA gastric cancer dataset. The patients were classified into RAB31-high and RAB31-low groups according to with median tpm of RAB31 across samples.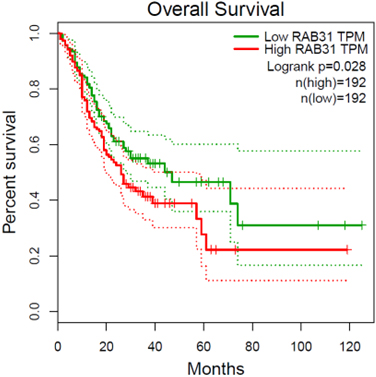
This study investigated the relationship between differentially expressed HERVs and protein-coding genes in gastric cancer. Our results support that HERVs act as a potential biomarker of gastric cancer.
To investigate the potential role of human endogenous retroviruses (HERV) in gastric cancer, we performed a differential analysis of HERV expression and developed SVM and AdaBoost models for multivariate screening to identify HERVs with major differences between tumor and normal groups. Our screen identified three key markers (LTR16A
To explore the relationship between HERVs and protein-coding genes, we divided the samples into HERV-high and HERV-low groups according to the median expression tpm of LTR16A
We observed that the well-known IL-17 pathway was the most significantly correlated functional modules, with a broad role in activating downstream signaling pathways involved in inflammatory responses, autoimmune diseases, and tumor development [35]. Additionally, the PI3K-Akt pathway showed consistent results with the pathway enrichment analysis [36]. Our analysis of HERV markers showed a significant correlation between inflammatory pathways and their expression changes. We propose that HERV may be activated by the inflammatory response, resulting in a significantly altered expression pattern during the inflammation-to-tumor process. The overexpression of HERV may affect the expression of downstream genes, leading to an overall acceleration of tumor development.
In this study, we utilized the WGCNA approach to analyze the correlation between HERV markers and protein-coding genes and performed a detailed functional analysis of HERV in tumorigenesis. We can predict the future survival status of patients through the changes in the levels of HERV markers in transcriptome sequencing results, and by further identifying the conditions of patients through these highly related genes and pathways, this provides a multi-dimensional assessment basis for the early screening of gastric cancer. The HERV markers and correlated genes may pave the way for experimental verification of their potential roles in tumor development.
Conclusion
Our study revealed a significant relationship between differentially expressed HERV and protein-coding genes in gastric cancer. The functional analysis supported that HERVs may be potential biomarkers of gastric cancer. Based on the differential analysis of HERV expression, we developed SVM and AdaBoost models, and identified three key HERV markers (LTR16A
Author contributions
Conception: Zhengtai Li and Changyuan Yu.
Interpretation or analysis of data: Zhengtai Li, Hongjiang Li.
Preparation of the manuscript: Zhengtai Li, Kun Fang and Xinglei Lin.
Revision for important intellectual content: Chang-yuan Yu.
Supervision: Changyuan Yu.
Data availability
The annotation file used for quantification and the quantification results can be found at the following URL:
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-230417.
Footnotes
Acknowledgments
National Natural Science Foundation of China (Grant No. 82174531); Shen Zhen Science and Technology Project (JCYJ20180507183842516).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.