
Abstract
BACKGROUND:
Colon adenocarcinoma (COAD) is a globally prevalent cancer, with hormone secretion playing a crucial role in its progression. Despite this, there is limited understanding of the impact of hormone secretion on COAD prognosis. This study aimed to establish a prognostic signature based on hormone secretion-related genes and to elucidate the potential functional mechanisms of these genes in COAD.
METHODS:
Using data from The Cancer Genome Atlas COAD cohort (TCGA-COAD), six hormone secretion-related genes were identified (CYP19A1, FOXD1, GRP, INHBB, SPP1, and UCN). These genes were used to develop a Hormone secretion score (HSS), which was then evaluated using the Kaplan-Meier curve and multivariable Cox analysis. The HSS model was further validated with external GEO cohorts (GSE41258, GSE39582, and GSE87211). Functional enrichment analyses were performed, and the CIBERSORT and TIDE algorithms were used to assess tumor infiltration.
RESULTS:
The study developed a prognostic signature, dividing patients into HSS-high and HSS-low groups. The HSS-high group showed a notably worse prognosis within the TCGA-COAD dataset and in three independent datasets: GSE41258, GSE39582, and GSE87211. Moreover, the HSS-high group predicted a shorter overall survival rate in patients maintaining microsatellite stability (MSS). The functional analysis associated HSS-high with the hypoxic, epithelial-mesenchymal transition (EMT), and TGF-
CONCLUSIONS:
This study demonstrated the prognostic significance of a HSS signature based on six hormone secretion-related genes in COAD. The findings suggest that this gene signature may serve as a reliable biomarker for predicting survival outcomes in COAD patients.
Introduction
Colorectal cancer (CRC), one of the most common malignant tumors globally, significantly impacts both men and women and serves as a leading contributor to cancer-related deaths in China [1, 2, 3]. Colon adenocarcinoma (COAD), the most prevalent histological subtype, constitutes approximately 90% of all CRC cases [4]. CRC displays considerable heterogeneity, as shown by the range of subtypes defined by anatomical location or molecular alterations [5]. An alarming rise in CRC incidence among young adults has been noted in recent years [6]. Despite substantial advancements in COAD treatments, including chemotherapy, radiation therapy, and immunotherapy. Over the past decades, the prognosis for patients with COAD, particularly those with lymph node metastases, remains poor [7, 8]. Therefore, the urgent and unfulfilled need to decode the molecular underpinnings of COAD’s pathogenesis remains central to identifying innovative therapeutic avenues and prognostic biomarkers.
The hormone secretion pathway holds significant sway in the development of cancer. Previously study indicated that hormones, which regulate the standard growth of target organs, can also foster conditions conducive to neoplastic transformation [9, 10]. Both endogenous and exogenous hormones stimulate cell proliferation, thereby elevating the likelihood of accumulating errors in random inheritance [11]. This process can lead to the onset of malignant phenotypes, as observed in various types of cancer such as breast [12, 13, 14], endometrial [15, 16], prostate [17, 18], and ovarian cancers [19, 20]. The significant advancements in hormone therapy have notably improved survival rates in the treatment of localized and metastatic breast, ovarian, endometrial, and prostate cancers [21].
Previous study has demonstrated that hormones also play a role in CRC [18, 22, 23]. For instance, Harbs et al. reported that elevated levels of circulating testosterone and sex hormone-binding globulin could heighten the risk of CRC in men [18]. Moreover, oral supplementation with the gut-derived peptide hormone uroguanylin has been found to inhibit polyp formation in mice and to suppress CRC proliferation without inducing serious side effects [22, 23]. However, the prognostic implications of hormones in CRC remain inadequately defined.
In this study, we leveraged public datasets to formulate and validate a COAD prognostic signature predicated on hormone secretion-related genes. Furthermore, we investigated the correlations between the devised hormone signature and clinicopathological characteristics. These findings could provide a fundamental basis for elucidating associations that are critical to the diagnosis and treatment of COAD, thereby influencing future strategic planning in this field.
Materials and methods
Data collection
Gene expression profiles of COAD and matched non-cancerous samples were downloaded from The Cancer Genome Atlas (TCGA,
Differentially expressed genes analysis
Expressions of 314 hormone secretion-related genes were extracted from the TCGA expression profile data, 66 genes were not found, and genes with expression greater than 0 in more than half of the samples were retained. Finally, 248 genes were included for subsequent analyses. The raw read counts were utilized to identify differentially expressed genes (DEG) between normal and tumor samples by R package “DEseq2”. The up-regulated and down-regulated gene was defined with a
Overall survival analysis and validation of candidate genes
To detect the prognosis-related genes in COAD, we used TPM and Kaplan – Meier curves (R package “survival” and “survminer”) to analyze the expression and prognosis value of 176 hormone secretion members in TCGA-COAD. Among them, six hormone secretion-related genes (CYT19A1, FOXD1, GRP, INHBB, SPP1, UCN) whose expression levels were related to the OS were further validated with GEO datasets. Finally, all these six genes were used to construct a prognostic signature for future analyses.
Calculation of gene signature score of the hormone secretion members
The hormone secretion signature (HSS) score was generated by summing the Z scores of six HSS genes as previously reported [17, 24, 25]. TCGA-COAD samples were grouped as either HSS-high (top 25% of HSS scores) or HSS-low [15, 26]. To identify the effect of the HSS score on the prognosis of COAD, Kaplan–Meier plotter was applied to explore the association of the HSS score with prognosis in TCGA-COAD patients.
Gene set enrichment analysis and Gene Ontology analysis
The gene sets of the latest Hallmark (v7.4) from the MsigDB database (
Multivariate cox analysis
Multivariate analysis with the Cox proportional hazards model was adopted to identify independent factors associated with OS in COAD. This analysis was performed with R packages (“survival”, “survminer” and “forestplot”).
Statistics analysis
Statistical analyses were conducted using the R platform (R v4.0.3) to assess significant differences. These assessments utilized a two-tailed Student’s
Workflow of this study. TCGA, The Cancer Genome Atlas; COAD, colon adenocarcinoma; DEG, differentially expressed genes; FC, fold change.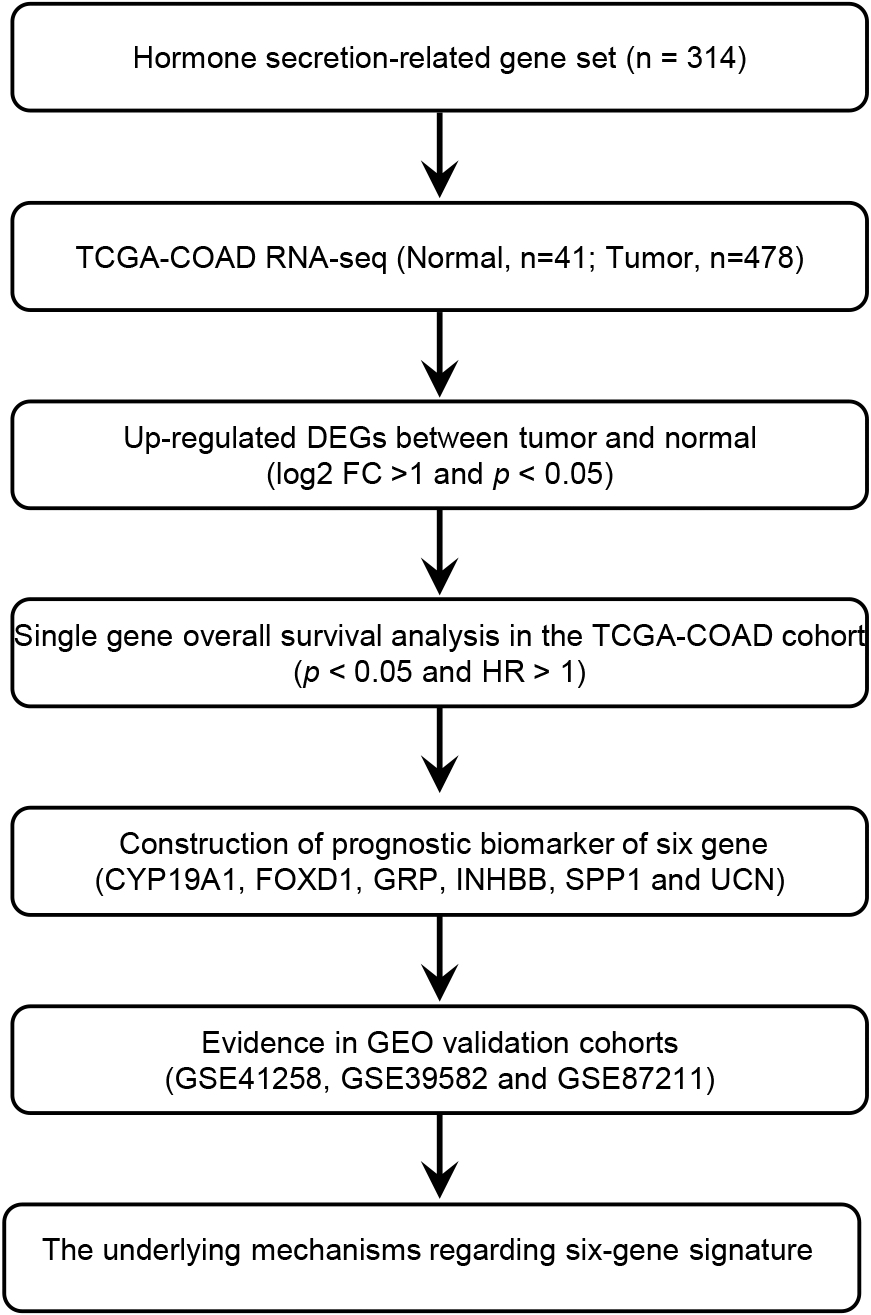
Forty-five hormone-related DEGs in COAD based on the TCGA-COAD dataset. TCGA, The Cancer Genome Atlas; COAD, colon adenocarcinoma; DEG, differentially expressed genes.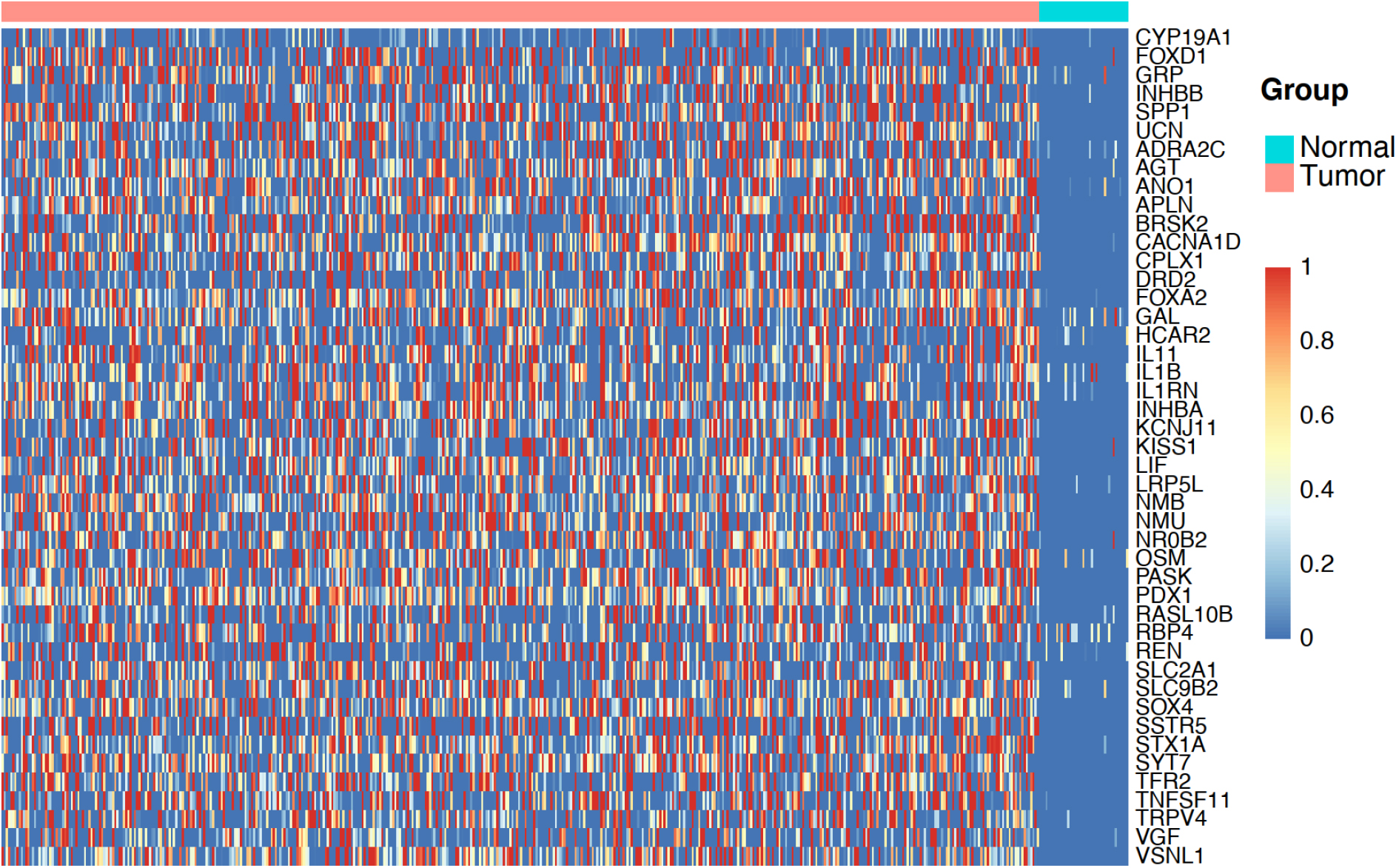
Screening of hormone secretion-related differentially expressed genes with prognostic significance from TCGA
This study utilized a total of 519 patients from The Cancer Genome Atlas colon adenocarcinoma (TCGA-COAD) dataset and three COAD validation datasets from the GEO database. The workflow is depicted in Fig. 1. The TCGA-COAD dataset contained 41 normal samples and 478 tumor samples. Due to the presence of multiple COAD samples from a single patient, 455 COAD patients were ultimately included in this study. By comparing COAD tumor samples with normal samples, 45 hormone secretion-related genes were identified as up-regulated differentially expressed genes (DEGs) (
Subsequently, Kaplan–Meier survival analysis was performed to identify DEGs significantly correlated to OS, 6 genes were risk factors with hazard ratios (HR)
Overall survival analysis based on the TCGA-COAD dataset. (A) Kaplan-Meier Curves indicating the associations of six hormone-related DEGs with overall survival in COAD. (B) The expression of six hormone related DEGs in tumor and normal samples. All 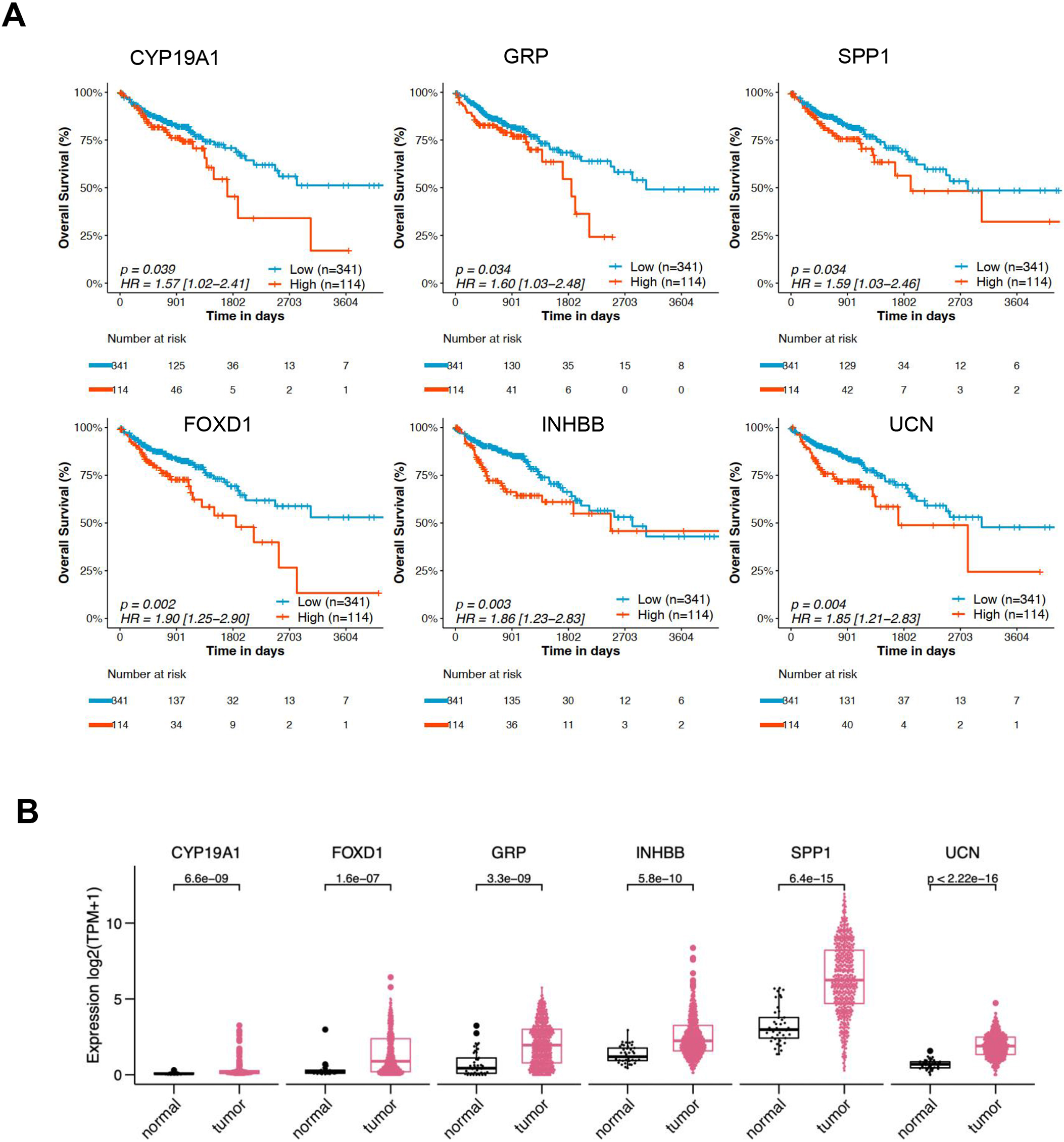
The six prognosis-related genes were subjected to z-score analysis. According to the sum of the
The prognostic value of six hormone-secretion related genes signature in the TCGA-COAD and GEO-COAD datasets. (A) Kaplan-Meier Curves indicating the associations of six hormone-secretion related genes signature (hormone signature score, HSS 25% vs. 75%) with overall survival in the TCGA-COAD dataset; (B) Kaplan-Meier Curves indicating the associations of the signature with overall survival in the GSE41258 dataset; (C) Kaplan-Meier Curves indicating the associations of the signature with overall survival in the GSE39582 dataset; (D) Kaplan-Meier Curves indicating the associations of the signature with overall survival in the GSE87211 dataset. TCGA, The Cancer Genome Atlas; COAD, colon adenocarcinoma; GEO, Gene Expression Omnibus. All 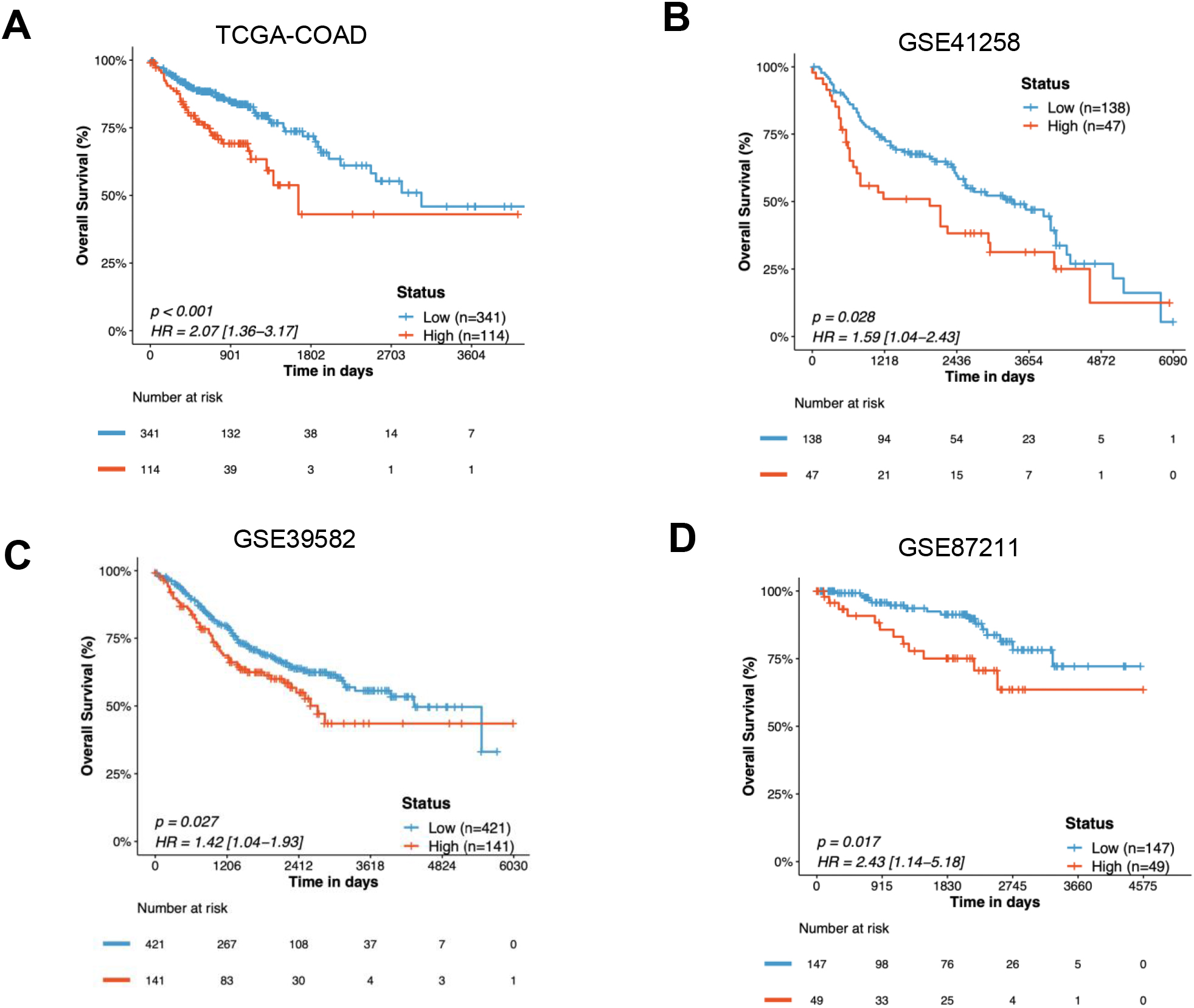
In addition, the data from GSE41258 including 185 COAD patients (
Multivariate analysis with the cox proportional hazards model on the independent factors associated with overall survival in the TCGA-COAD dataset. TCGA, The Cancer Genome Atlas; COAD, colon adenocarcinoma; MSI-H, microsatellite instability-high; MSS, microsatellite stability. All 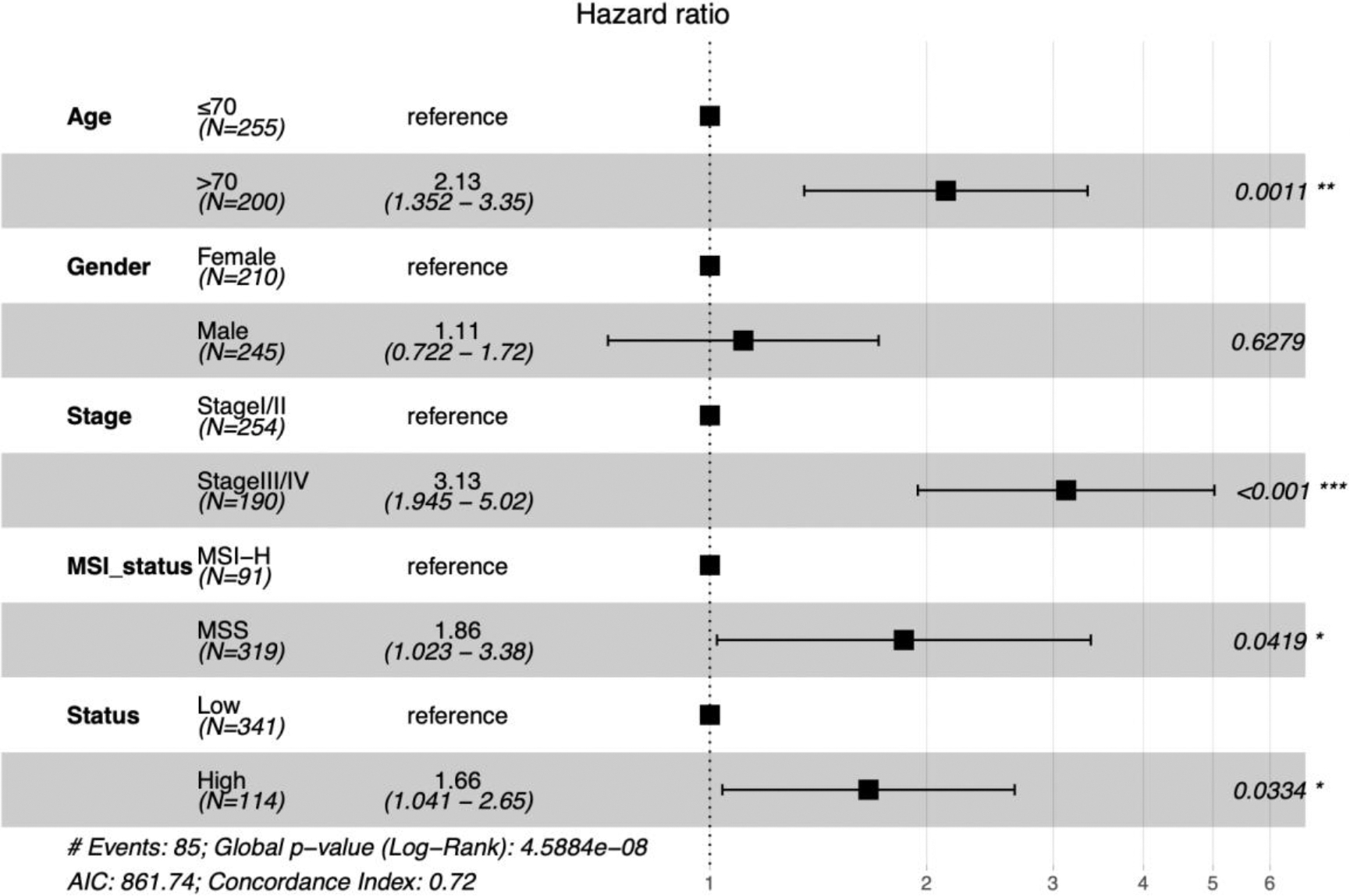
Next, we performed Hallmark enrichment analysis using the gene sets from MSigDB between the HSS-high and HSS-low score groups (Fig. 6A). Given the results, we observed that the hypoxic pathway (Fig. 6A) and EMT pathway (Fig. 6A) were significantly enriched in the HSS-high group. In addition, the KEGG analysis supported that the HSS score was positively correlated with the transforming growth factor-
Functional enrichment analysis of the HSS-high group. (A) Hallmark enrichment analysis of DEGs identified in HSS-high vs. HSS-low group; (B) KEGG enrichment analysis of DEGs identified in HSS-high vs. HSS-low group; (C) the difference of EMT score between HSS-high and HSS-low group calculated using the GSVA algorithm; (D) the difference of TGF-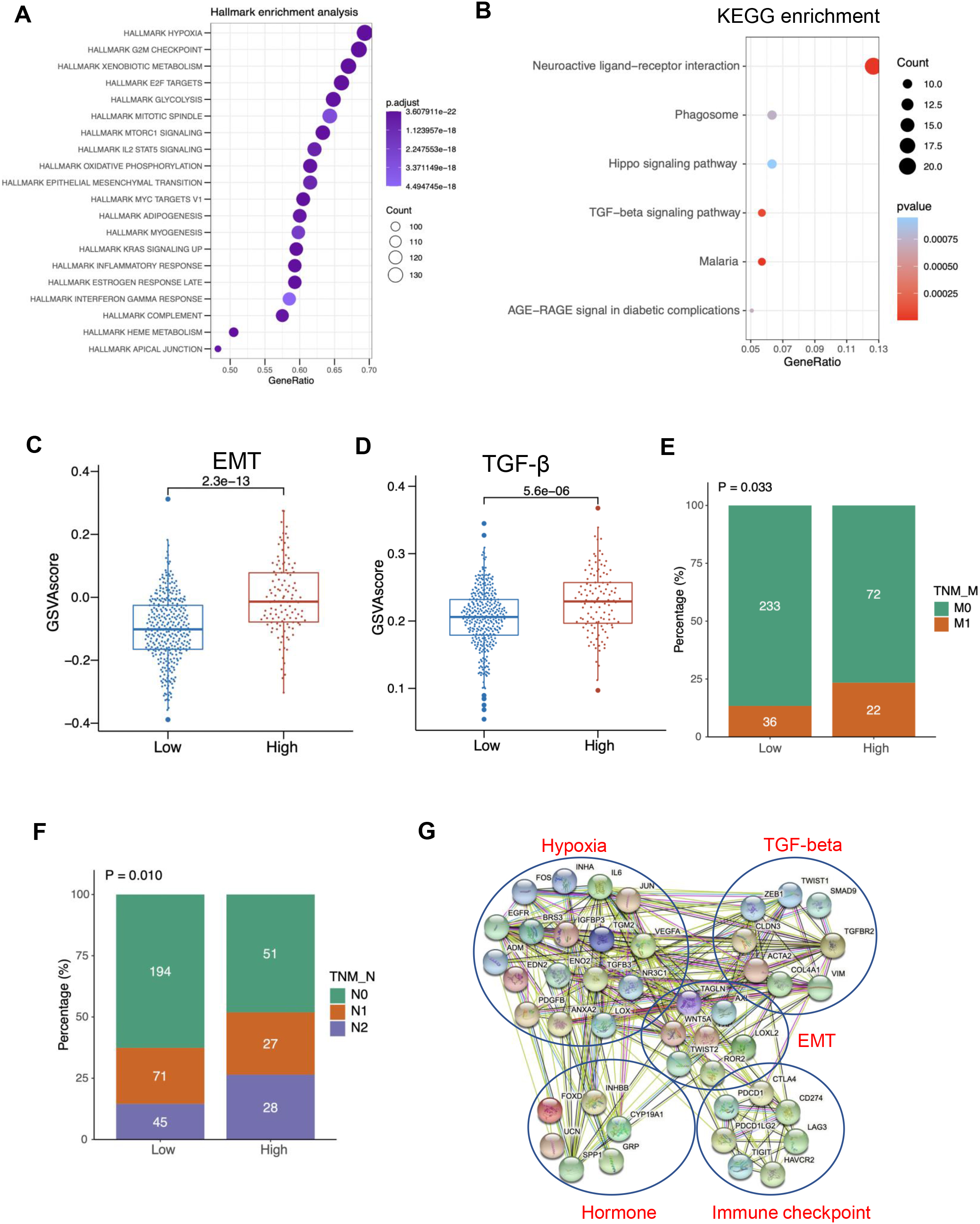
To further uncover the correlations between HSS score and related pathways, we also conducted a GSEA analysis (Figure S3D-3F). Encouragingly, the results showed significant enrichment of the hypoxic pathway, EMT pathway, and estrogen response pathway in the HSS-high group, proving that this six-gene prognostic signature could predict the status in COAD patients.
EMT is a cellular process implicated in cancer metastasis, while the TGF-
Tumor-infiltrating analysis between the HSS-high and HSS-low score groups
Tumor microenvironment analysis. (A) CIBERSORT analysis in the HSS-high and HSS-low groups; (B) TIDE analysis in the HSS-high and HSS-low groups. All 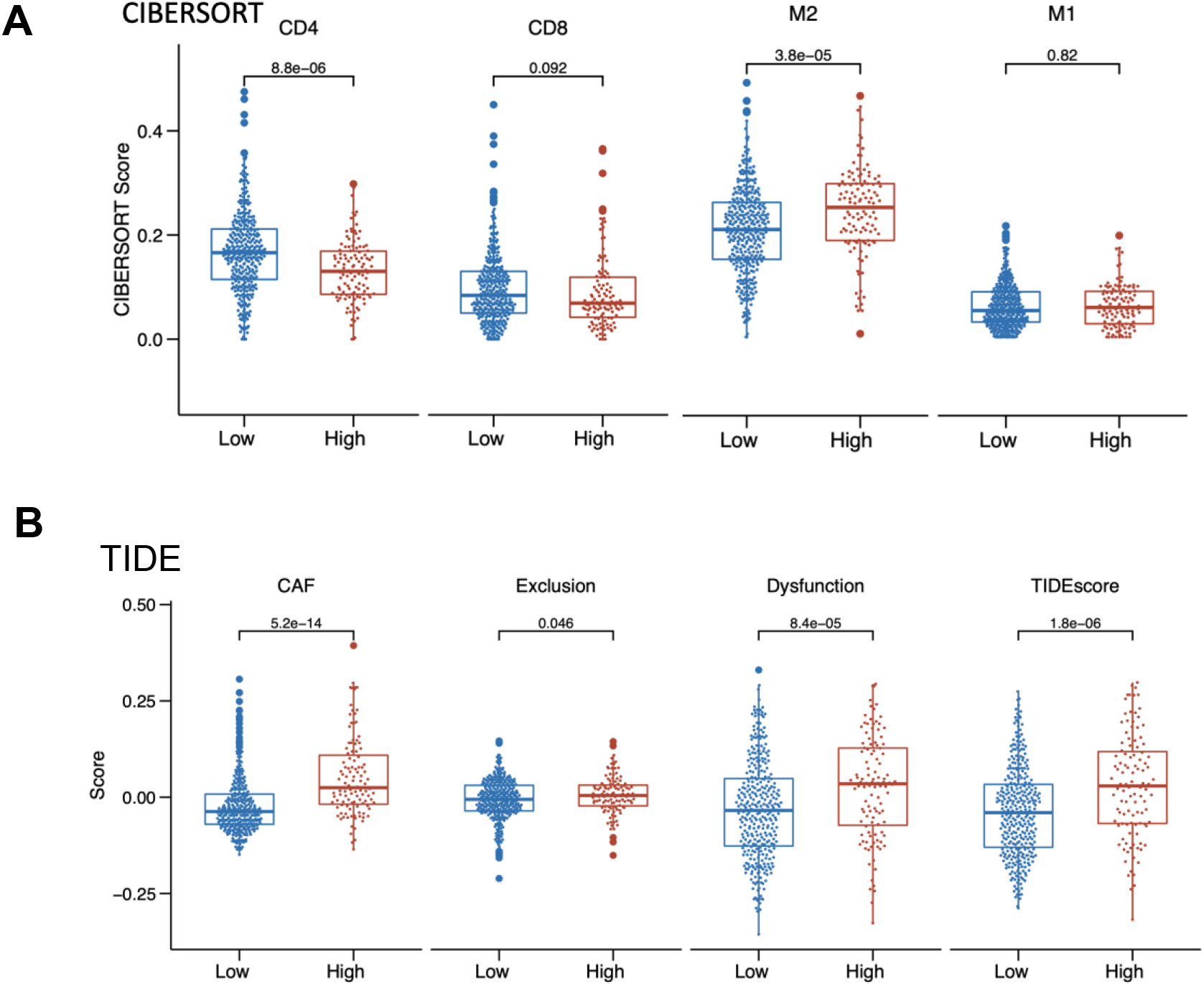
Subsequent analyses assessed the influence of the HSS score on the tumor microenvironment. The CIBERSORT algorithm was utilized to estimate the proportions of tumor-infiltrating immune cells in COAD patients. The analysis showed that the group with a high HSS score (HSS-high) had a lower proportion of CD4 and CD8 cells than the group with a low HSS score (HSS-low), as depicted in Fig. 7A. Additionally, the proportion of M2 macrophages was significantly higher in the HSS-high group, suggesting a correlation between a high HSS score and tumor metastasis. Using the Tumor Immune Dysfunction and Exclusion (TIDE) algorithm, the potential clinical efficacy of immunotherapy in different HSS score groups was examined. A higher TIDE prediction score suggests an increased likelihood of immune evasion and a reduced likelihood of benefiting from immune checkpoint inhibitor (ICI) treatment. Results indicated that the HSS-high group had a higher TIDE score than the HSS-low group, suggesting that patients with a high HSS score are less likely to benefit from ICI treatment (Fig. 7B). Furthermore, the HSS-high group had a higher cancer-associated fibroblast (CAF) score and increased T cell exclusion and dysfunction scores (Fig. 7B). These findings suggest that the HSS-low group, with its higher levels of tumor-infiltrating cells, might be more likely to benefit from ICI treatment.
Despite the progress in therapeutic approaches, there is still a lack of effective prognostic indicators [27]. The utilization of hormone therapy in breast and prostate cancers has changed the prognosis and treatment of patients, however, the link between CRC and hormones is less well known. In this study, the six hormone secretion gene signature (CYT19A1, FOXD1, GRP, INHBB, SPP1, and UCN) was constructed. The associations of the signature with survival outcomes in COAD were explored. Moreover, the potential molecular mechanisms of the signature implicated in COAD tumorigenesis were also investigated based on public datasets.
The CYP19A1 is recognized as an influential lipid metabolism-related gene (LMG) in colon cancer, as revealed in the previously described [28]. The study aimed to create a lipid metabolism-related prognostic risk score (LMrisk) and utilized differentially expressed LMGs, including CYP19A1. The CYP19A1 was identified as an independent prognostic factor and was found to be positively correlated with PD-L1 expression in human colon cancer tissues. Inhibition of CYP19A1 was observed to enhance CD8+ T cell-mediated antitumor immune response, induce normalization of tumor blood vessels, and boost the efficacy of anti-PD-1 therapy in colon cancer models. This was achieved through the downregulation of PD-L1, IL-6 and TGF-
FOXD1, or foxhead box D1, has been identified as a critical player in various aspects of CRC. According to several studies, FOXD1 is involved in the mechanism of CXCL5’s role in tumor angiogenesis in CRC. Its influence is traced to the activation of the AKT/NF-
The gastrin-releasing peptide (GRP) and its receptor (GRPR) play significant roles in the development and progression of CRC. Notably, both amidated and non-amidated forms of GRP were found to accelerate CRC cell proliferation and migration, mediated through the GRP receptor. These effects were inhibited by a GRP receptor antagonist, indicating the potential therapeutic applications for GRP receptor antagonists in CRC management [31]. GRPR is ectopically expressed in over 60% of colon cancers, and its expression is correlated with increased colon cancer cell migration. Upon activation, GRP stimulates the small GTPase RhoA through G13 heterotrimeric G-protein signaling. The regulator of G-protein signaling, postsynaptic density 95/disk-large/ZO-1 (PDZ)-RhoGEF (PRG), has been identified as the main activator of RhoA downstream of GRPR [32]. This PRG-RhoA-ROCK pathway contributes to GRP-stimulated colon cancer cell migration and Cox-2 expression, which in turn increases prostaglandin-E2 (PGE2) production, further enhancing cell migration. GRP and GRPR can also act as morphogens in CRC, influencing cell motility and enhancing cell-matrix attachment, processes mediated by focal adhesion kinase (FAK) [33]. Interestingly, GRP-induced FAK activation upregulates the expression of ICAM-1, a molecule implicated in enhancing cell motility and attachment to the extracellular matrix. Therefore, GRP is impact on CRC cell behavior and progression involves multiple molecular pathways and interactions.
Inhibin subunit beta B (INHBB), a protein-coding gene involved in the synthesis of the transforming growth factor-
Secreted phosphoprotein 1 (SPP1), also known as osteopontin, plays a significant role in CRC. In multiple studies, SPP1 was found to be overexpressed in CRC tissues, correlating with invasion and metastasis. In the tumor microenvironment, SPP1 enriched macrophages were found to interact with fibroblast activation protein (FAP)+ fibroblasts, potentially contributing to the formation of an immune-excluded structure that inhibits T cell infiltration. Inhibiting this interaction could potentially improve the efficacy of immunotherapy [35, 36]. Furthermore, a study demonstrated that overexpression of SPP1 can promote the process known as epithelial-mesenchymal-transition (EMT), which is associated with increased cancer cell invasiveness and metastasis [37]. Lastly, a seven-gene prognostic model, including SPP1, was constructed and validated, capable of predicting disease-specific survival in CRC patients [28]. This model also revealed an immunosuppressive tumor microenvironment in high-risk CRC patients. The presence of SPP1+ macrophages, found to be highly expressed in a subset of macrophages with strong senescence-associated secretory phenotype (SASP) features, was linked with poor prognosis in CRC. Overall, these findings indicate that SPP1 could serve as a potential therapeutic target in CRC.
Urocortin (UCN) is a part of several immune gene models which have been developed to predict the prognosis of COAD and colon cancer. In one study, UCN was identified as one of twelve immune-related genes that were used to construct a risk model for colon cancer [28]. This model was verified as an independent prognostic variable with good prognostic ability, and the increase in risk score was associated with a rise in macrophages, myeloid dendritic cells, and CD4+ T cells. Another study identified UCN as one of seven immune genes that can be used to form a model for predicting the prognosis of COAD, potentially providing potential therapeutic targets for clinical treatment [38]. These studies suggest that UCN plays a significant role in the immune response within the tumor microenvironment of COAD and colon cancer, and could be a valuable biomarker for prognosis and treatment planning.
This study reveals a significant increase in the expression of six hormone secretion-related genes in COAD tumor samples compared to normal tissue. By summing the z-score of each gene, a hormone secretion score (HSS) is computed. Analysis of COAD datasets from TCGA and three GEO repositories indicates that patients with a high HSS suffer from significantly shorter overall survival than those with a low HSS. Further exploration of the molecular mechanisms related to these six hormone secretion genes in COAD identifies several hallmark pathways. These pathways, significantly enriched in the high HSS group compared to the low HSS group, include the hypoxia pathway, the TGF-
Hypoxia, a defining characteristic in CRC development [39], triggers the expression of genes linked to inflammatory response, tumor vascularization, and metastasis by stabilizing the HIF1a transcription factor [1, 38, 40]. The EMT is another crucial physiological process pivotal to embryonic development and wound healing [41]. It’s also a notable feature associated with cancer invasion and metastasis [42, 43]. Previous studies have revealed a significant link between the upregulation of SPP1, a gene enriched in the EMT pathway, and enhanced cell growth, adherence, and invasion in CRC [37, 42]. Functioning as a cytokine, SPP1 amplifies the production of interferon-gamma (IFN-
Recent research reveals that CRC exhibits significant stromal TGF-
These findings suggest that the six identified hormone-secretion-related genes contribute to the progression and metastasis of COAD. Specifically, HSS-high could potentially intensify the EMT process via the activation of the TGF-
However, the study has certain limitations. First, the conclusions are largely based on bioinformatics analysis and need further verification through in vivo and in vitro studies. Second, the study does not incorporate key risk factors for CRC, such as dietary habits and family history.
In conclusion, this study identified a signature of six hormone-secretion-related genes that influence the TGF-
Author contribution
Conception: Yongkun Sun, Yin Guan.
Interpretation or analysis of data: Xiongjie Jia, Tao Zhang, Xinze Lv, Haiwei Du, Yongkun Sun, Yin Guan.
Preparation of the manuscript: Xiongjie Jia, Tao Zhang, Xinze Lv, Haiwei Du.
Revision for important intellectual content: Yongkun Sun, Yin Guan.
Supervision: Yongkun Sun, Yin Guan.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-230126.
sj-xlsx-1-cbm-10.3233_CBM-230126.xlsx - Supplemental material
Supplemental material, sj-xlsx-1-cbm-10.3233_CBM-230126.xlsx
Footnotes
Conflict of interest
Xinze Lv and Haiwei Du were employed by the company Burning Rock Biotech Ltd. at the time of writing this report. The other authors have no conflicts of interest to declare.