
Abstract
OBJECTIVE:
Testicular germ cell tumors (TGCTs), containing pure seminoma and non-seminoma, occupy the most majority of testicular cancers in adolescents and young men, which has increased dramatically in recent decades. Therefore, it is important to find crucial genes for improving diagnosis and prognosis in TGCTs. However, the diagnostic and prognostic markers of TGCTs are limited.
METHODS:
In this study, our main objective is to explore novel potential genes that can be used as diagnostic and prognostic biomarkers in TGCTs. Our study detected 732 differentially expressed genes (DEGs) using three microarray expression profiling datasets from Gene Expression Omnibus (GEO). Multiple analysis was performed to identify the roles of DEGs, including pathway and functional enrichment analysis, protein-protein interaction (PPI) network analysis, module analysis, and survival analysis.
RESULT:
In total, 322 upregulated genes and 406 downregulated genes were identified as DEGs The functional and pathway enrichment analysis shows that DEGs were highly enriched in multiple biological attributes such as T cell activation, reproduction in multicellular organism, sperm flagellum, antigen processing and presentation Then, seven potential crucial genes were identified via PPI network analysis, module analysis, and survival analysis. Furthermore, 7 potential crucial genes had shown to play a key role in regulating immune cell infiltration level in patients with TGCTs.
CONCLUSION:
We identified seven potential crucial genes (LAPTM5, NCF2, PECAM1, CD14, COL4A2, ANPEP and RGS1), which may be molecular markers in improving the way of diagnosis and prognosis in TGCTs.
Introduction
Details testicular germ cell tumors datasets downloaded from GEO
Details testicular germ cell tumors datasets downloaded from GEO
In recent decades, testicular germ cell tumors(TGCTs) patients have increased dramatically, and the age distribution is mainly between 15 and 35 years [1]. TGCTs are divided into two main categories: pure seminoma and nonseminoma Non-seminoma could also be divided into four subcategories: embryonal carcinoma, yolk sac tumor, choriocarcinoma and teratoma [2]. In different ethnic groups, the incidence rate of TGCTs has obvious diversity, which indicates that the interaction of inheritance and environment are involved in the pathogenesis of TGCTs, but the specific mechanisms of the occurrence and development of TGCTs still need to be explored [3]. TGCTs are considered treatable even in disseminated disease [4]. However, the treatment outcome of low-risk patients is poor, and some metastatic patients will not be able to receive first-line treatment based on cisplatin and salvage treatment [5, 6]. The leading cause of these adverse events is that the diagnosis and prognosis markers of TGCTs are still incompletely clear. Therefore the objective of this study is to explore and identify potential and valuable diagnostic and prognostic markers for patients with TGCTs. Human chorionic gonadotrophin (hCG) and alpha-fetoprotein (AFP) are often used as diagnostic and prognostic markers in serum from patients with TGCTs. However, hCG has about a quarter of the opposite result hCG and AFP decreased or unchanged in patients with seminomas and moreover, about 30% of non-seminomas are negative [7]. The normal or abnormal markers level cannot diagnose all types of TGCTs and distinguish them from other cancers or some benign conditions, although they are both widely used in clinical practice for TGCTs patients [8].
The testis is an immune-privileged site, which can protect testicular tissue from autoimmune responses [9, 10, 11]. Testicular tissue was used to believe completely immune-privileged while many studies showed that the blood-testis barrier (BTB) could allow some testicular antigens to enter testicular tissue when the BTB was disrupted [12, 13, 14, 15]. Recent studies have reported that immune cell infiltration of TGCTs is related with tumor prognosis [16, 17, 18, 19]. Although there are many new therapeutic methods for targeted or biological treatments in TGCTs, the options available to these patients are limited [20, 21, 22, 23].
The purpose of this study is to determine the potential crucial genes for TGCTs through the gene expression omnibus (GEO) database, which will help to understand the potential mechanism for the occurrence and development of TGCTs. This research used bioinformatic methods, including pathway and functional enrichment analysis, PPI network analysis, module analysis, and survival analysis, to investigate the potential molecular mechanism of potential crucial genes in the pathogenesis and/or clinical prognosis of TGCTs. Our results in this research will help to improve and perfect our understanding of the occurrence and development of TGCTs and offer a novel insight into the treatment and diagnosis of TGCTs.
Datasets of testicular germ cell tumors
Three registered microarray datasets, including GSE3218, GSE8607, and GSE18155, were downloaded from the GEO database (
Volcano and heat maps of DEGs identified from 3 datasets.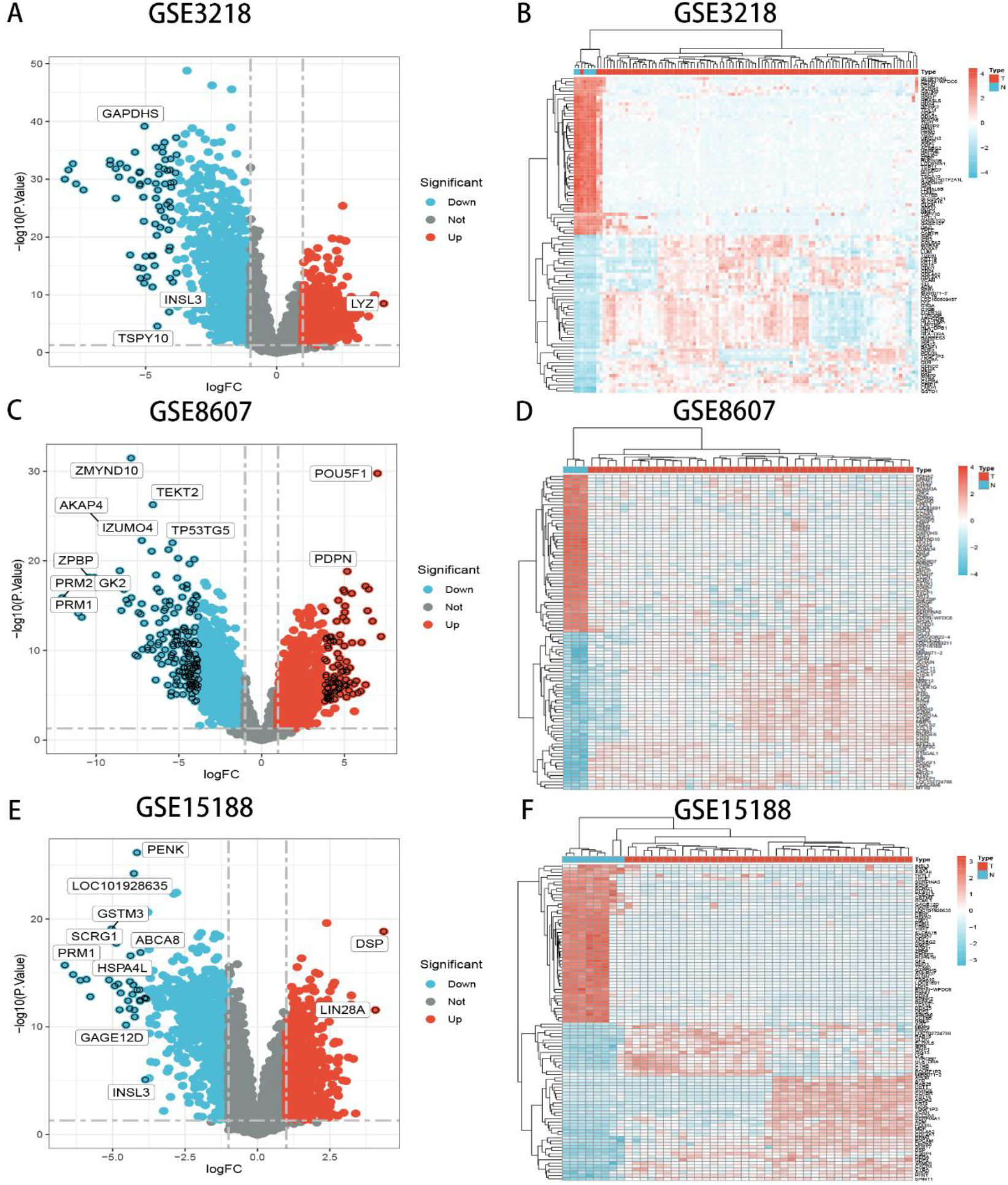
Venn plot and GO analysis of TGCTs. (A) 732 DEGs were identified by integrating 3 TGCTs datasets. (B–E) Results of the GO enrichment analysis of the DEGs that were common upregulated and downregulated genes.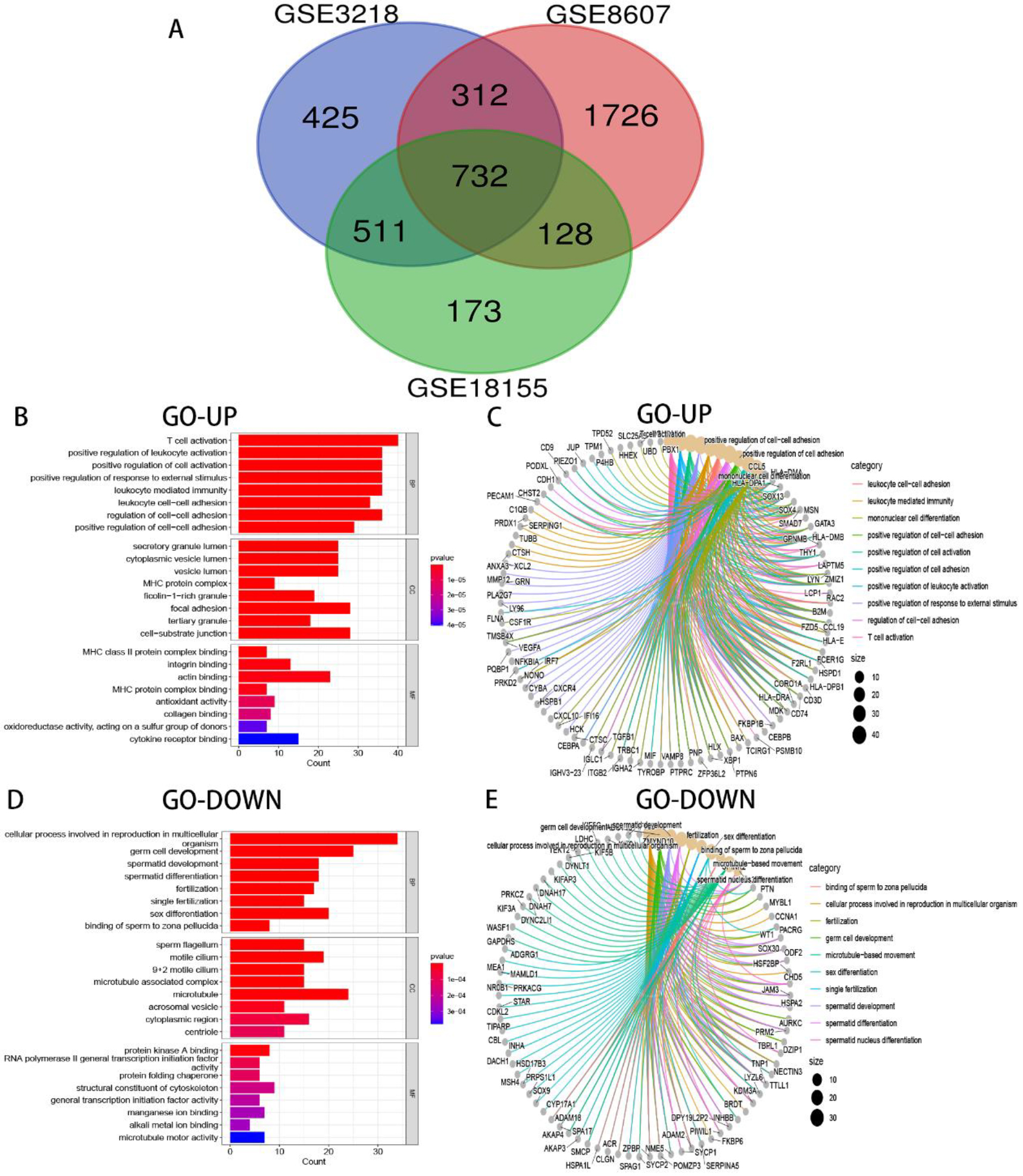
The “normalizeBetweenArrays” function of the limma package (
GO and KEGG analysis for DEGs
For the purpose of identification of the potential functional and pathway related to DEGs, Gene Ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were completed through the clusterProfiler package (
PPI network analysis and MCODE analysis
The correlations of common DEGs were constructed using STRING online software (
Survival analysis and protein atlas of potential crucial genes
The GEPIA2 database (
Correlations between immune cell infiltration and potential crucial gene expression based on TIMER
In order to explore the association between immune cell infiltration and potential crucial gene expression, we selected TIMER (
Results
732 DEGs were obtained through integrating 3 datasets. Among them, 322 genes were upregulated and 406 were downregulated (here we only list the top 10 significant DEGs respectively)
732 DEGs were obtained through integrating 3 datasets. Among them, 322 genes were upregulated and 406 were downregulated (here we only list the top 10 significant DEGs respectively)
The GEO datasets were searched for mRNA microarray data of tumor and normal tissue from studies relevant to TGCTs. Finally, we found three microarray expression datasets, including GSE3218, GSE8607 and GSE18155. 819 upregulated genes and 1161 downregulated genes were identified from 1980 DEGs of the GSE3218 dataset (Fig. 1A–B). In 2898 DEGs of the GSE8607 dataset, 1438 upregulated genes and 1460 downregulated genes were identified (Fig. 1C–D). 681 upregulated genes and 863 downregulated genes were identified from the GSE18155 dataset (Fig. 1E–F). The bioinformatics & evolutionary genomics was selected to draw Venn diagrams of DEGs of 3 datasets, and 732 DEGs were confirmed (Fig. 2A, Table 2). After integrating DEGs identified from 3 datasets, 322 upregulated genes and 406 downregulated genes which had the same trend, were identified from 3 datasets.
Functional and pathway enrichment analysis of DEGs with the same trend
Pathway enrichment analysis of upregulated and downregulated genes in TGCTs.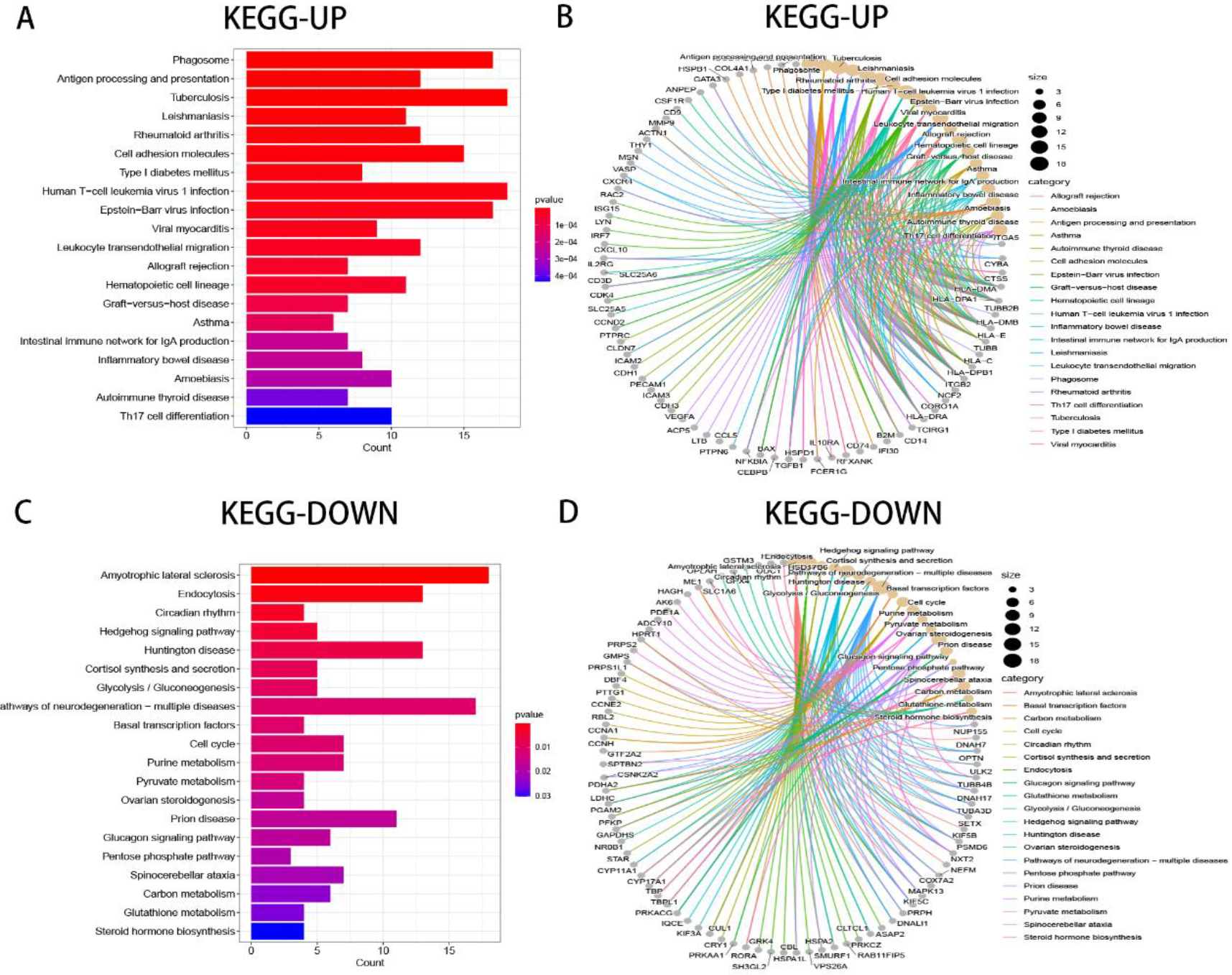
To further analyze the potential function of DEGs with the same trend, the R package of clusterProfiler was used to perform GO terms and KEGG pathways analysis. In the biological process (BP), upregulated genes were mainly enriched in T cell activation, positive regulation of leukocyte activation, and positive regulation of cell activation (Fig. 2B–C). On the contrary, downregulated genes were mostly enriched in cellular processes involved in reproduction in multicellular organism, germ cell development, and sex differentiation (Fig. 2D–E). In the cellular component (CC), upregulated genes were concentrated mainly in the secretory granule lumen, cytoplasmic vesicle lumen, and vesicle lumen (Fig. 2B–C). On the contrary, downregulated genes were mostly concentrated in the sperm flagellum, motile cilium, and 9
MCODE analysis in DEGs with the same trend. Upregulated genes are represented by red, and downregulated genes are represented by green.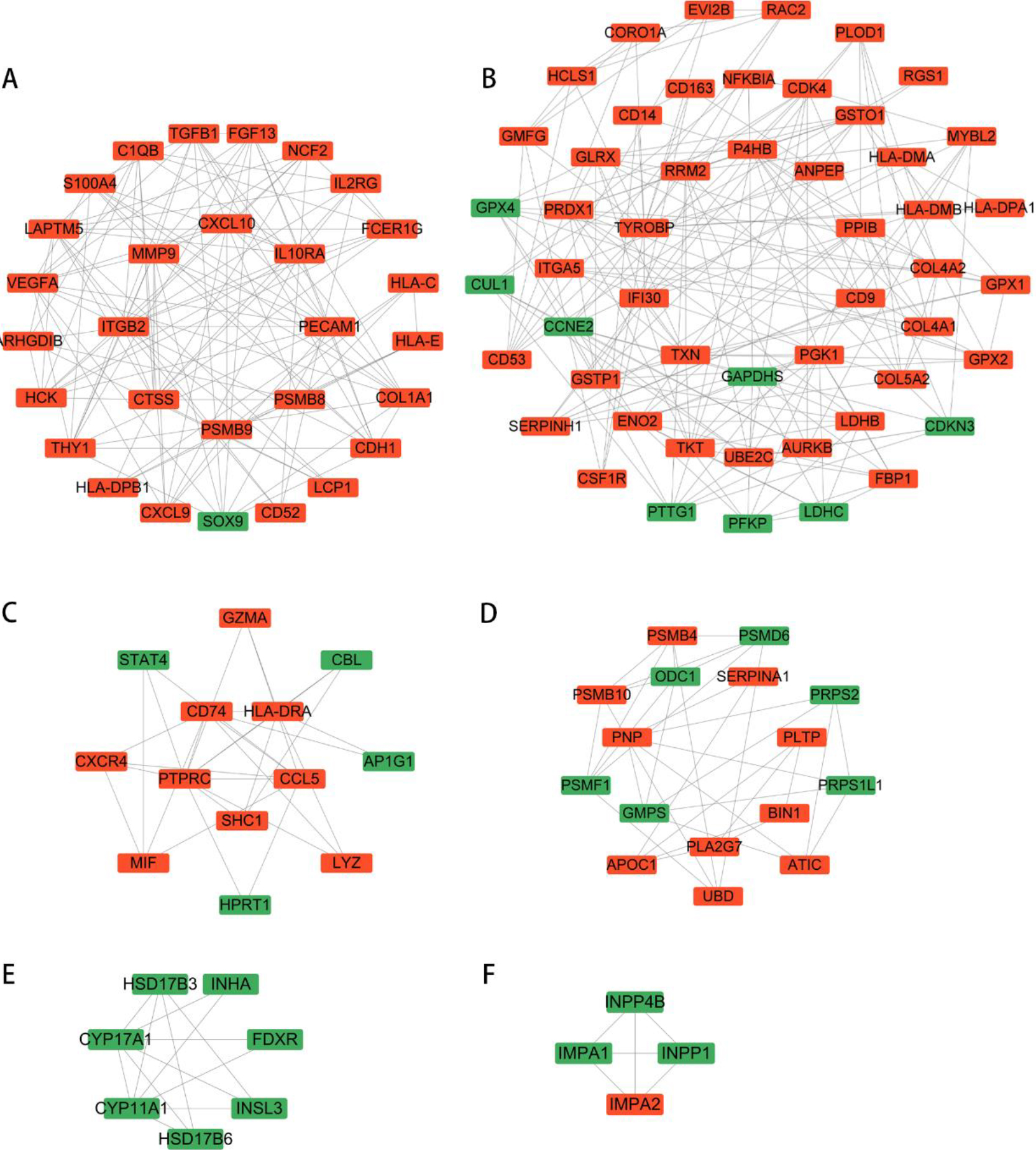
The STRING was selected to form a PPI network, consisting of 653 nodes and 3228 edges, and then used cytoscape analysis software to analyze this network. Consequently, six significant clustering modules were obtained using the function of MCODE plug-in of cytoscape (Fig. 4). Clustering module 1 was composed of 29 nodes and 122 edges (Fig. 4A). Clustering module 2 contained 51 nodes and 185 edges (Fig. 4B). Clustering module 3 included 13 nodes and 29 edges (Fig. 4C). Clustering module 4 included 16 nodes and 33 edges (Fig. 4D). Clustering module 5, included 7 nodes and 13 edges (Fig. 4E). Clustering module 6, composed of 4 nodes and 6 edges (Fig. 4F).
Association between the mRNA expression level of 7 potential crucial genes and the prognosis in TGCTs. (A–G) Interaction between mRNA expression and survival based on the GEPIA database. (H) Bar graph of GO analysis, colored by 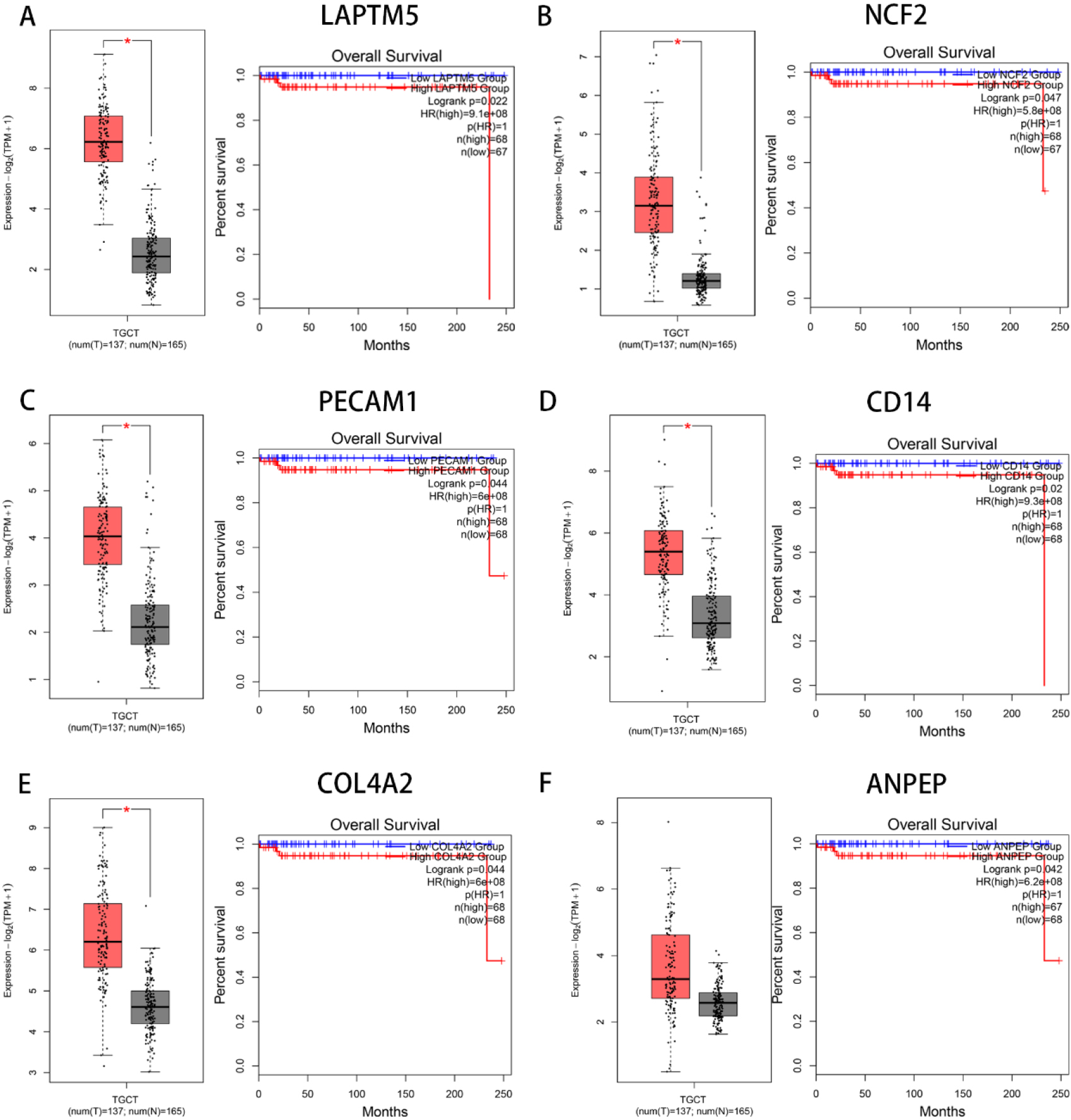
The mRNA expression level of 7 potential crucial genes in TGCTs tissue was higher than that in normal tissue in the HPA.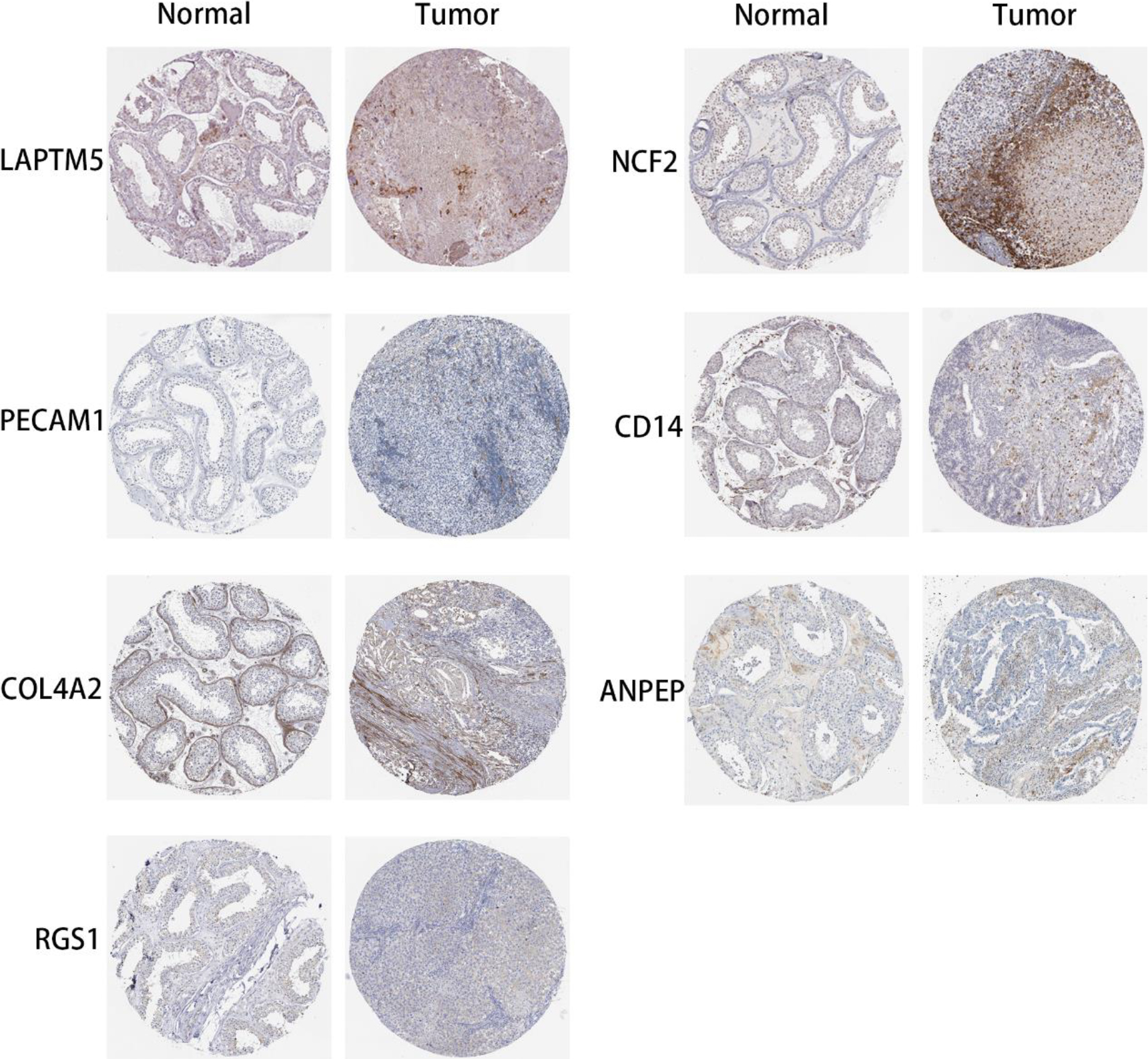
The prognostic evaluation of the hub genes obtained from the clustering module via MCODE analysis was evaluated using GEPIA. Survival analysis had shown that most hub genes were not relevant to the overall survival rate (OS) of patients with TGCTs. Despite what happened, we identified 7 genes (LAPTM5, NCF2, PECAM1, CD14, COL4A2, ANPEP and RGS1) in the end that were significantly associated with the OS rate of TGCTs patients, and 5 genes (LAPTM5, NCF2, PECAM1, CD14, COL4A2) were significantly associated with mRNA expression between the tumor and normal tissue (Fig. 5A–G). Using the Metascape database, it had shown that 7 genes were enriched in phagocytosis, neutrophil degranulation, and blood vessel development in the GO terms, and were concentrated mainly in the localization and development process in the biological process group (Fig. 5H I). Similarly, the expression of 7 potential crucial genes is upregulated in the tissue of patients with TGCTs comparied with the normal tissue in HPA (Fig. 6). Thus, we further explore them as potential crucial genes.
Correlation between the mRNA expressions level of 7 potential crucial genes and immune cell infiltration levels in TGCTs. (A–G) Correlation between the mRNA expressions level of 7 potential crucial genes and immune cell infiltration levels of B cell, CD4+ T cells, CD8+ T cells, macrophages, neutrophils, and dendritic cells. (H) Kaplan-Meier plots of immune cell infiltration in TGCTs. 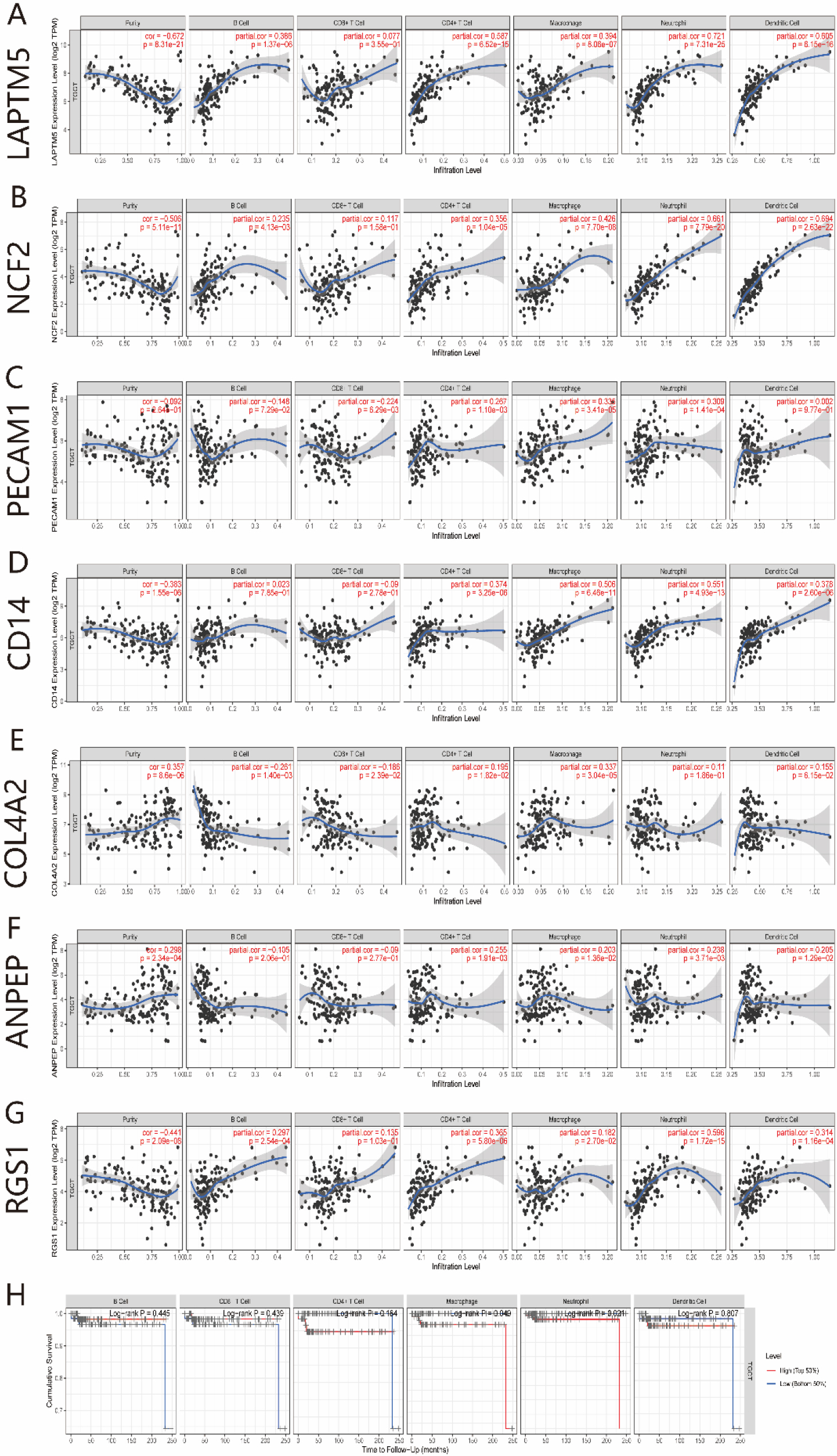
As the major elements of the tumor microenvironment, tumor-infiltrating lymphocytes can affect the initiation, progression and/or metastasis in various cancers. Therefore, we further analyzed the association between immune cell infiltration and the mRNA expression of potential crucial genes using TIMER We also analyzed the relationship between prognosis and immune cell infiltration in patients with TGCTs The results displayed that the mRNA expression level of genes, including LAPTM5, NCF2, PECAM1, CD14, COL4A2, ANPEP, and RGS1, had obvious correlations with immune cell infiltrating levels of B cell, CD4+ T cells, CD8+ T cells, Macrophages, Neutrophils, and Dendritic cells in TGCTs (Fig. 7A–G). Furthermore, the results showed that macrophage and neutrophil infiltration was significantly correlated with the of prognosis TGCTs (Fig. 7H). This suggests that 7 potential crucial genes may play a key role in regulating immune cell infiltration level in patients with TGCTs, and a particular effect on macrophage and neutrophils infiltration.
Discussion
Although the development of new technical products in diagnosis and treatment has improved survival rates of patients with TGCTs, and up to 95% of patients did not recur within 5 years, patients who received medical treatment may therefore have with adverse reproduction effects [24, 25, 26]. The long-term relative survival rate after diagnosis with TGCTs usually decreases continuously with the increasing of follow-up time, especially over 15–30 years [27]. Stroke, secondary leukemia, cerebrovascular accidents, kidney disease and internal carotid artery occlusion had been shown to be related to patients with TGCTs after chemotherapy in the literature [28].
In this study, we independently analyzed by comparing normal and tumor samples to identify potential pathogenesis genes in TGCTs. First, DEGs of three datasets were obtained, respectively using R packages. Then the results were integrated from 3 datasets and 732 DEGs with the same trend were confirmed including 322 upregulated genes and 406 downregulated genes. It is worth analyzing the potential roles of these DEGs in the occurrence and development of TGCTs. We also performed function and pathway enrichment analysis by using the clusterprofiler package to explore the regulatory processes and function of these DEGs The biological functions of DEGs mainly involve T cell activation, cellular process involved in reproduction in multicellular organism, and germ cell development. The pathways of the DEGs mainly involve Phagosome, Antigen processing and presentation and Circadian rhythm.
We identified hub genes from common DEGs through MCODE analysis and seven genes (LAPTM5, NCF2, PECAM1, CD14, COL4A2, ANPEP and RGS1) were identified as survival-related genes, which had not been explored by researchers in the studies of TGCTs before Therefore, experimental and clinical studies of 7 potential crucial genes in TGCTs need to be carried out by further studies. This discovery can provide the diagnostic and prognostic markers in TGCTs. Lysosomal-associated transmembrane protein 5 (LAPTM5) is a kind of membrane proteins expressed on the intracellular vesicles [29], having a close interaction with a variety of human cancers, such as glioblastoma, bladder cancer, estrogen receptorpositive (ER+) breast cancer, neuroblastomas [30, 31, 32, 33]. However, the connection between LAPTM5 and TGCTs remains largely unknown. Neutrophil cytoplasmic factor 2 (NCF2) is the component of cytosolic NADPH oxidase, which involves gastric cancer angiogenesis and metastasis through the LINC01410-miR-532-NCF2-NF-kB pathway [34]. Platelet EC adhesion molecule (PECAM1, also known as CD31) also has been associated with multiple cancers [35, 36, 37], however, there are few reports of TGCTs. The C-terminal portion of collagen type IV alpha 2 (COL4A2) had been reported to inhibit tumor growth, which is the main components of basement membrane [38]. Recently, the crucial role of COL4A2 in epithelial ovarian cancer was reported to promote anoikis resistance through upregulation [39]. Aminopeptidase N (APN, also known as CD13; encoded by ANPEP) is an adverse prognostic marker for prostate cancer (PC), and an epigenetic mechanism is involved in the inhibition of ANPEP in PC [40]. Upregulation of regulator of G protein signaling 1 (RGS 1) in tumor-specific T cells could reduce the survival time of patients with breast or lung cancer, indicating that RGS1 may offer a new immunotherapy strategy for patients with TGCTs [41]. Human monocyte differentiation antigen CD14 is a pattern recognition receptor (PRR), which was first confirmed as a monocyte marker of intracellular responses after bacterial infection and can enhance innate immune responses [42]. CD14-high bladder cancer cells may exacerbate the inflammatory response and accelerate tumor growth through enhanced tumor cell proliferation [43].
Tumor-infiltrating lymphocytes as the major components of the tumor microenvironment, affect the initiation, progression and/or metastasis of various tumor patients [44, 45]. Therefore, we further analyzed the relationship between potential crucial genes and immune infiltration. Our analytical results showed that the mRNA expression level of 7 potential crucial genes has a significant association with the infiltration levels of B cell, CD4+ T cells, CD8+ T cells, macrophages, neutrophils, and dendritic cells in TGCTs. Further analysis showed that macrophage and neutrophil infiltration significantly correlated with TGCTs prognosis. Immune infiltration has been reported in the development of TGCTs, while the molecular mechanisms responsible remain unknown [46, 47, 48, 49]. Interestingly, there are few reports on potential crucial genes in TGCTs. Therefore, our study may improve novel targets in immune infiltration for TGCTs and novel diagnostic tools.
Conclusion
We used multiple analysis to identify DEGs from 3 datasets and found seven novel survival-related genes (LAPTM5, NCF2, PECAM1, CD14, COL4A2, ANPEP and RGS1), providing potential diagnostic and prognostic markers for patients with TGCTs. In addition, more future scientific research needs to be carried out to study the association between 7 potential crucial genes and TGCTs. Because of limitations in our study design, further epidemiological and experimental studies is required for explore the association between seven novel genes and TGCTs
Statement of ethics
Due to the nature of this study, no ethical approval was required.
Funding sources
This work was supported by National Natural Science Foundation of China (No. 81302452), Natural Science Foundation of Jiangsu Province (BK20221374), Major Projects of Natural Sciences of University in Jiangsu Province of China (18KJB330004), a project fund of Basic Scientific Research program of Nantong City (JC2019021, JC2020032), an innovation project of graduate student scientific research in Jiangsu province (KYCX22_3377) and Scientific Research Starting Foundation for The Doctoral researcher of Nantong University (No. 14B14).
Author contributions
Data analysis, interpretation and writing of this man-uscript: Shaokai Zheng. Conceptualization, methodology and supervision: Ting Li. Reviewing and editing the manuscript: Lianglin Qiu. All authors have seen and approved the final manuscript.
Data availability statement
The data sets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Footnotes
Acknowledgments
We acknowledge the TIMER, TCGA, GEPIA, GEO, STRING, human protein atlas and other databases for their selfless givenness.
Conflict of interest
The authors declare no conflict of interest.