
Abstract
BACKGROUND:
Liver hepatocellular carcinoma (LIHC) is one of the most malignancy over the world. Previous studies have proven that Molecules Interacting with CasL-Like 1 (MICALL1) participated in cellular trafficking cascades, while there has no study to explore the function and carcinogenic mechanism MICALL1 in LIHC.
METHODS:
We aimed to investigate the relationship between MICALL1 mRNA expression and LIHC using TCGA database. The expression of MICALL1 protein in clinic samples were examined by UALCAN database. Kaplan-Meier method was used for survival analysis. Logistic regression and Cox regression were performed to evaluate the prognostic significance of MICALL1. The MICALL1-binding protein were built by the STRING tool. Enrichment analysis by GO, KEGG and GSEA was used to explore possible function of MICALL1. The ssGSEA method was used to investigate the association between MICALL1 expression and the immune infiltration level in LIHC.
RESULTS:
The expression and prognostic value of different MICAL family members in LIHC were evaluated. The expression of MICALL1 was significantly increased at both the transcript and protein levels in LIHC tissues. Further, the LIHC patients with high MICALL1 levels showed a worse OS, DSS and PFI. Some clinicopathologic features were identified to be related to MICALL1 expression in LIHC included clinical T stage, pathologic stage, histologic grade and AFP concentration. Univariate and multivariate survival analysis showed that MICALL1 was an independent prognostic marker for OS and DSS. Further enrichment analysis revealed that the K-RAS, TNF
CONCLUSIONS:
MICALL1 expression was significantly associated with immune cell infiltration and may regarded as a promising prognostic biomarker for LIHC patients.
Background
Liver hepatocellular carcinoma (LIHC) is the most common type of liver cancer and is one of the most lethal malignancies worldwide [1]. A global toll of 906,000 cases and 830,000 death of liver cancer have been reported to occur during 2020 [2]. At present, the clinical treatment of LIHC includes surgical resection, radiofrequency ablation, neoadjuvant chemotherapy, and liver transplantation, etc [3, 4, 5, 6]. Despite a variety of treatments, there is a lack of a cure for LIHC. Therefore, there is an urgent need for new and more reliable biomarkers for the diagnosis, prognosis evaluation and targeted therapy of this disease.
It has been proven that cytoskeleton dynamics is closely related to LIHC development, metastasis, and recurrence [7, 8]. This cytoskeleton remodeling supports specific demands for mechano-signal transduction and motility of tumor cells. It also contributes to the progression of hepatitis [9, 10, 11], which is one of the leading cause of LIHC. Interference with cytoskeletal remodeling may be an important strategy for preventing dissemination of tumor cells. For instance, paclitaxel, a disruptor of microtubule cytoskeleton, is an effective drug for multiple tumor suppression. Molecules Interacting with CasLs (MICALs) is a newly found group of endogenous enzyme that involves in oxidization of actin and remodeling of actin cytoskeleton [12]. However, the role of MICALs in the prognosis of LIHC and its possible pathogenesis are still unclear.
While there is only one gene encoding MICAL in Drosophila, vertebrates contain five genes encoding MICAL isoforms (MICAL1-3) and MICAL-like isoforms (MICALL1-L2). MICALs are multidomain proteins and members of MICAL family share similar structures [13, 14]. Although the role of MICALL1 in carcinogenesis is unclear till now, evidence has indicated that all the other MICALs are participate in multiple tumors’ progression [15, 16, 17, 18, 19]. Some researchers argue that the depletion of MICALL1 inhibits cell surface delivery of TNF
In this study, we first identified the transcriptional and protein expression of MICALL1 via The Cancer Genome Atlas (TCGA), Alabama Cancer Database (UALCAN) and Human Protein Atlas (HPA) databases. Then, we predict biological function of MICALL1 by Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene set enrichment analysis (GSEA). Furthermore, we analyzed clinical features, immune cell filtration and prognostic values of MICALL1 in LIHC. The current study shows MICALL1 was a reliable and promising prognostic biomarker for LIHC, which will be beneficial to the diagnosis and treatment of LIHC.
Methods
Patients in TCGA database
Normalized RNA-seq data and relevant clinical information of 374 LIHC tumor tissues and 50 normal tissues were downloaded from TCGA database (
By the median of mRNA expression, we divided the LIHC patients into MICALL1-high and MICALL1-low expression groups. The data were gathered and analyzed via R3.6.3 software [28]. The association between MICALL1 mRNA expression and the OS, DSS and PFI of patients with LIHC were calculated by using the TCGA-LIHC dataset.
Kaplan-Meier plotter database
The prognostic value of MICAL family members in LIHC was assessed according to OS using Kaplan-Meier plotter (kmplot.com/analysis) [29].
Hepatocellular carcinoma database
HCCDB (
Protein expression database
Comparisons of MICALL1 expression between normal tissues and LIHC tissues were analyzed by HPA database (
GO, KEGG and GSEA analysis
The enriched pathways were collected using Gene Ontology (GO) [31, 32], KEGG [33, 34, 35] and GSEA [36, 37]. MICALL1 co-expression was analyzed statistically using Pearson’s correlation coefficient and performs analysis of Gene Ontology biological process (GO_BP), Gene Ontology cellular component (GO_CC), Gene Ontology molecular function (GO_MF), KEGG pathways by LinkedOmics database (
Protein-protein interaction (PPI) network analysis
In order to establish the interaction relationship between MICALL1 and its upstream and downstream proteins in LIHC, we used the STRING database (
Immune cells infiltration of ssGSEA
Immune infiltration of LIHC was identified by single-sample gene set enrichment analysis (ssGSEA) [37, 40]. The correlation analysis between MICALL1 and the infiltration levels of these 24 immune cell types were quantified by Spearman correlation test.
Statistical analysis
Univariate and multivariate analysis were used to assess the influence of clinical variables on survival. Statistical analysis was done with R software and SPSS 22.0 software.
Results
Expression of MICALL1 in patients with LIHC
First, we evaluated the expression and prognostic role of each MICAL family member in LIHC. All the five family members, MICAL1-3 and MICALL1-2 were found to be significantly highly expressed in LIHC (Fig. 1A). Among them, MICALL1 was not only significantly highly expressed in LIHC, but also associated with poor OS of LIHC patients (Fig. 1B). Additionally, the expression of MICALL1 was significantly higher in 50 LIHC samples when compared with that in matched adjacent samples (Fig. 1C). So, MICALL1 was screened out among the five genes in LIHC We noticed that the protein expression level of MICALL1 was higher in LIHC tissues in comparison with normal tissues (Fig. 1D), indicating that both mRNA and protein expressions of MICALL1 were significantly high expressed in patients with LIHC. The Receiver Operating Characteristic (ROC) analysis results also revealed that MICALL1 had high diagnostic potential, with the area under the curve (AUC) values of 0.895 (Fig. 1E).
The expression of MICALs in LIHC tissues. (A) The mRNA expression of individual MICAL members in LIHC tissues. (B) Kaplan-Meier analysis of OS for MICAL members in LIHC patients. (C) The mRNA expressions of MICALL1 in LIHC samples and paired adjacent samples. (D) MICALL1 protein levels in LIHC tissues (left). Representative images of MICALL1 expression in LIHC from HPA (right). (E) The ROC curve for MICALL1 show promising discrimination power between normal and LIHC samples. (F) Differences of MICALL1 expressions between tumor samples and normal samples from the GTEx combined adjacent normal tissues. (G) Chart and plot showing the expression of MICALL1 in hepatocellular carcinoma (HCC) tissues and the adjacent normal tissues from HCCDB.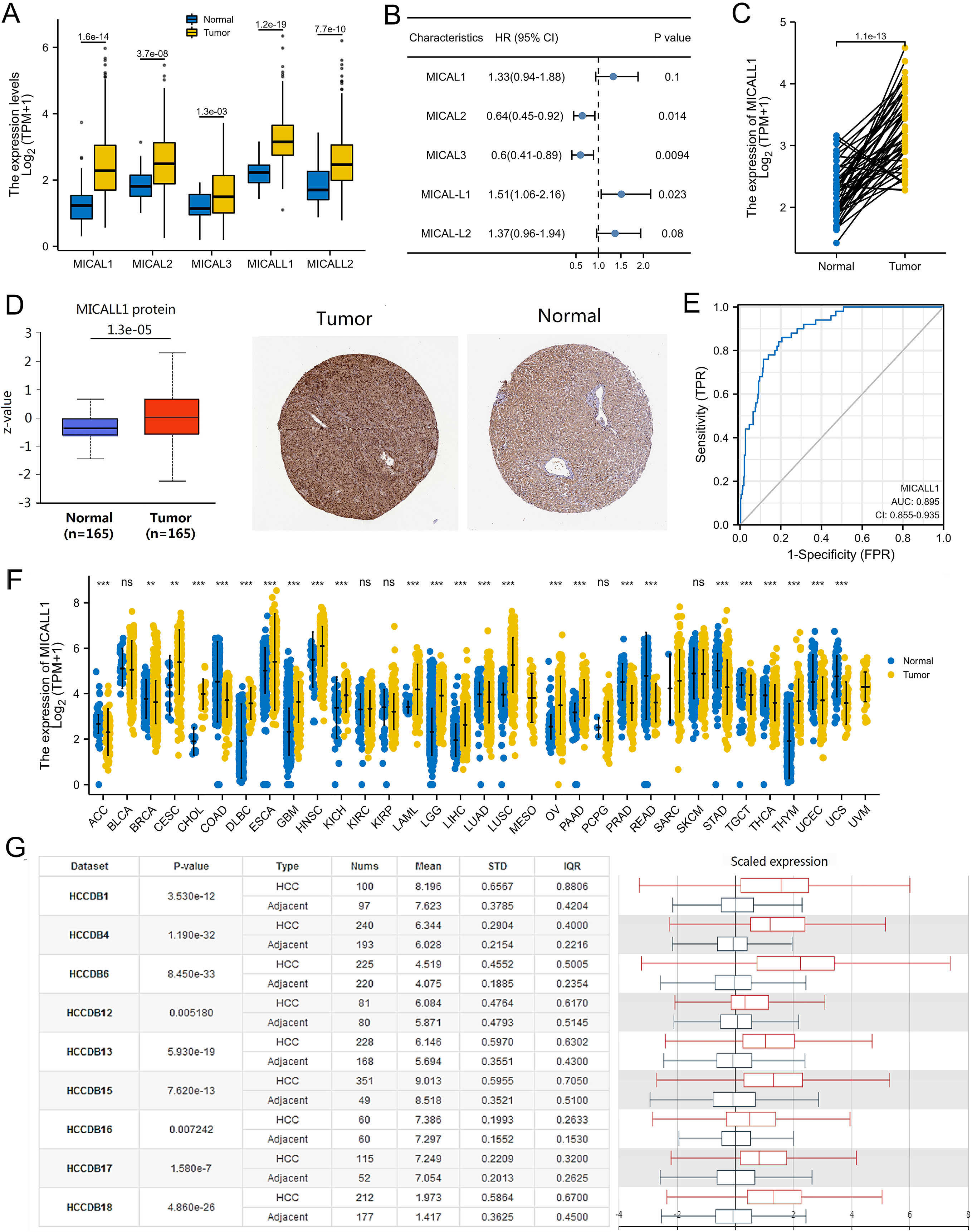
We investigated the expressions of MICALL1 in pan-cancer and found that mRNA expression of MICALL1 was significantly up-regulated in multiple types of cancers (Fig. 1F). To further explore the association between MICALL1 and LIHC, we analyzed of nine HCC cohorts in the HCCDB database and revealed that mRNA expression of MICALL1 was significantly higher in HCC tissues than in adjacent normal tissues (Fig. 1G).
Among the 374 LIHC patients, 187 (50.0%) were in the MICALL1 low expression group and 187 (50.0%) in the high MICALL1 expression group. The comparison of baseline data between the two groups revealed that MICALL1 expression was significantly associated with AFP concentration (
The relationship between MICALL1 expression and clinicopathological data
The relationship between MICALL1 expression and clinicopathological data
Association between MICALL1 expression and clinicopathologic characteristics (logistic regression analysis)
Logistic regression analysis demonstrated that MICALL1 expression was correlated with some clinical characteristics in patients with LIHC (Table 2). A comparison of baseline data between the MICALL1 expression groups and clinical characteristics using logistic regression analysis revealed that MICALL1 expression was significantly associated with T stage (T2&T3&T4 vs. T1) (OR
Univariate and multivariate Cox proportional hazards analysis of the correlation between MICALL1 expression and OS for LIHC patients
Univariate and multivariate Cox proportional hazards analysis of the correlation between MICALL1 expression and DSS for LIHC patients
Box plot assessing MICALL1 expression in patients with LIHC according to different clinical characteristics. (A) BMI, (B) Gender, (C) Age, (D) AFP, (E) T stage, (F) Pathologic stage, (G) Histologic grade.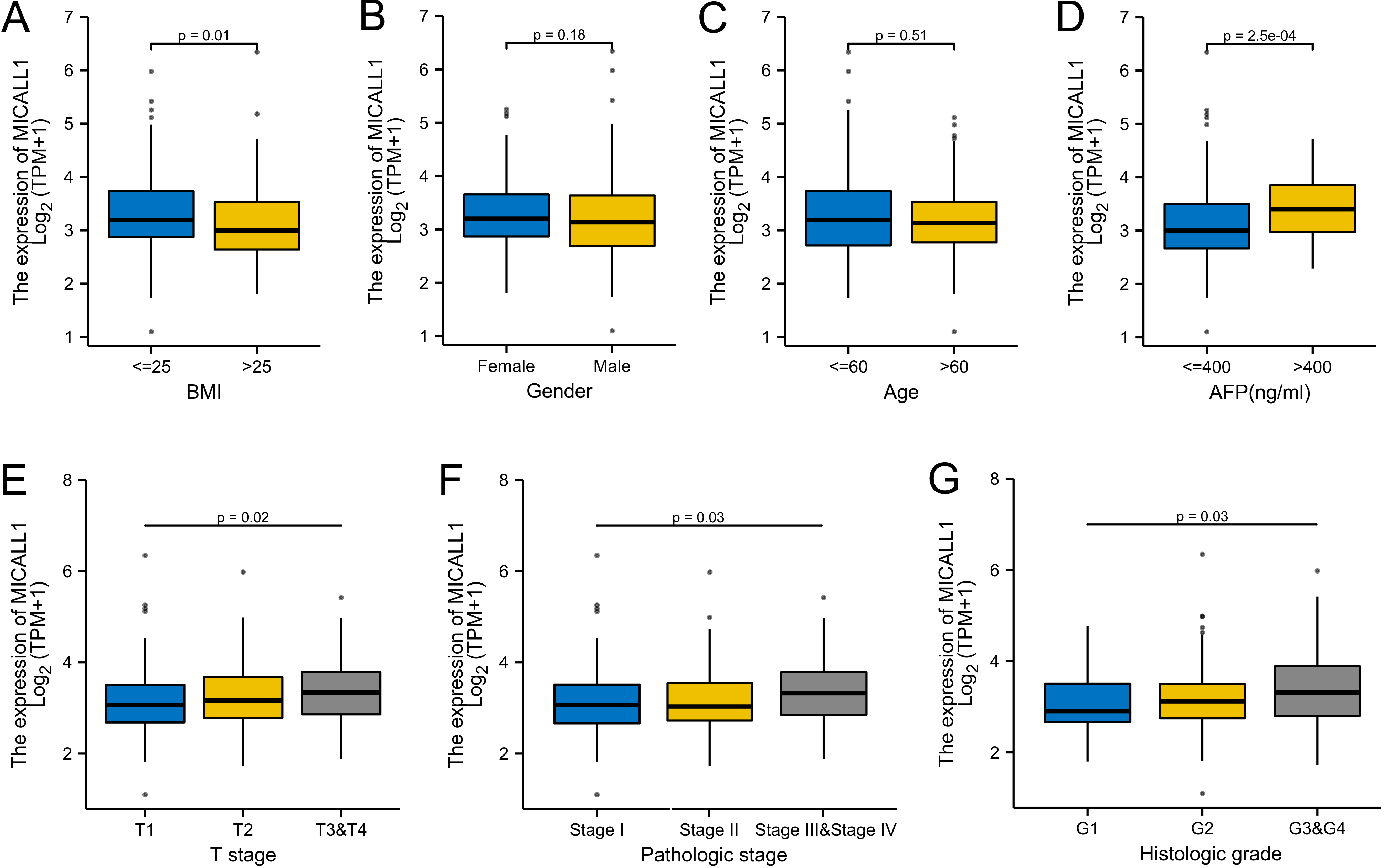
Prognostic value of MICALL1 expression for clinical outcomes in LIHC patients. Kaplan-Meier analysis of (A) OS, (B) DSS, (C) PFI for patients with LIHC. (D–K) Kaplan-Meier analysis of subgroup OS in patients. (D) Age: 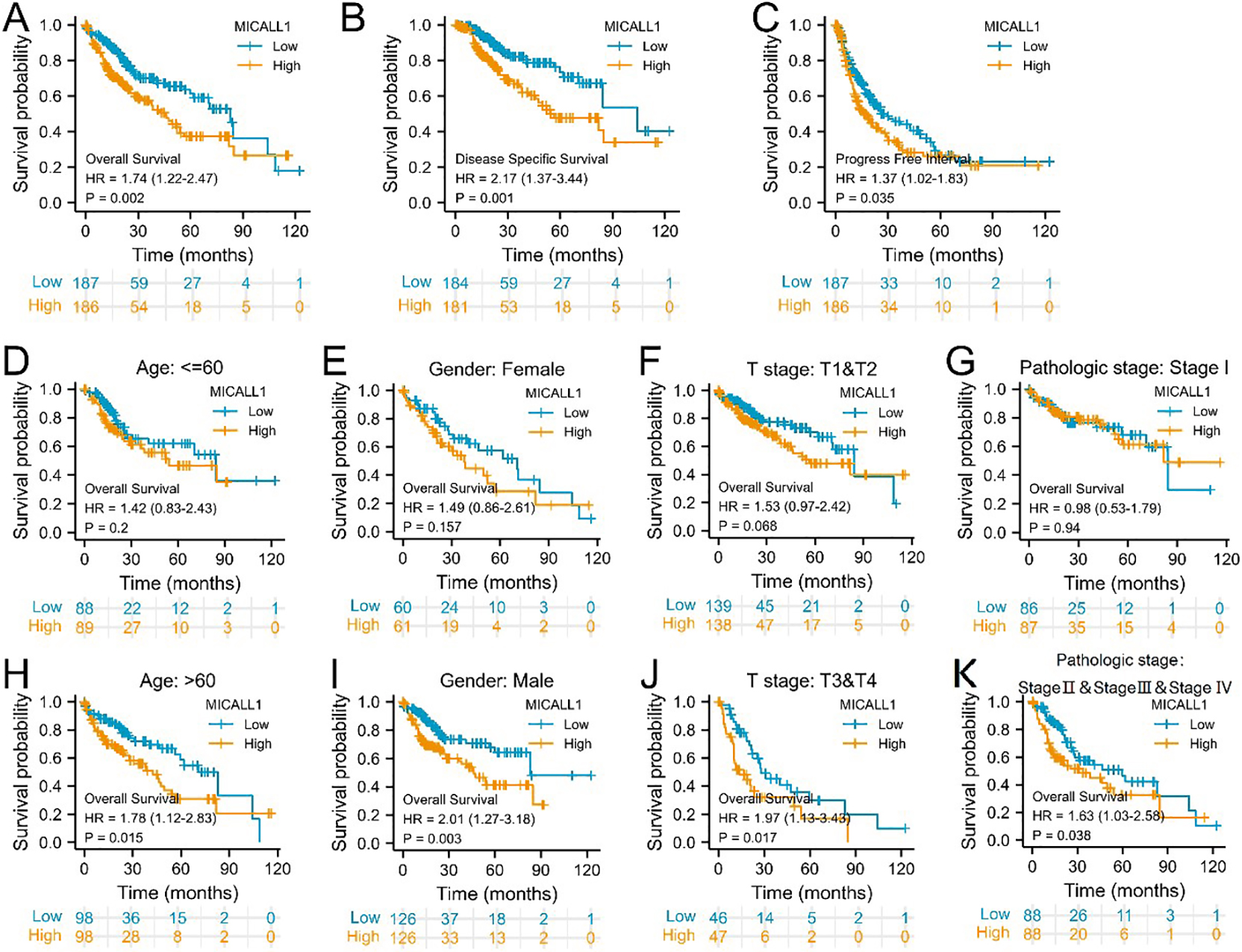
Then, whether MICALL1 expression is associated with LIHC outcome was determined. Survival analysis indicated that high MICALL1 expression was correlated with poor OS (HR
Univariate Cox regression analysis showed that high MICALL1 expression was significantly correlated with poor OS (HR
Establishing protein interaction networks
Using text mining and experimental evidence identification, the MICALL1-binding protein interaction network was established and visualized by STRING analysis (Fig. 4A). Moreover, by comparing MICALL1-interacted genes and the DEGs with MICALL1 expression in LIHC (Fig. 4B), a common member Rab36 was screened out (Fig. 4C). Furthermore, there was a remarkable positive association between MICALL1 mRNA expression and that of Rab36 (
An intersection analysis of MICALL1-interacted genes and DEGs of MICALL1. (A) The interaction network of MICALL1-binding proteins was obtained from the STRING database. (B) Gene expression differences was visualized by a volcanic map. (C) An intersection analysis of MICALL1-interacted genes and DEGs of MICALL1 was performed. (D) Correlation analysis between MICALL1 expression and screened out common gene Rab36.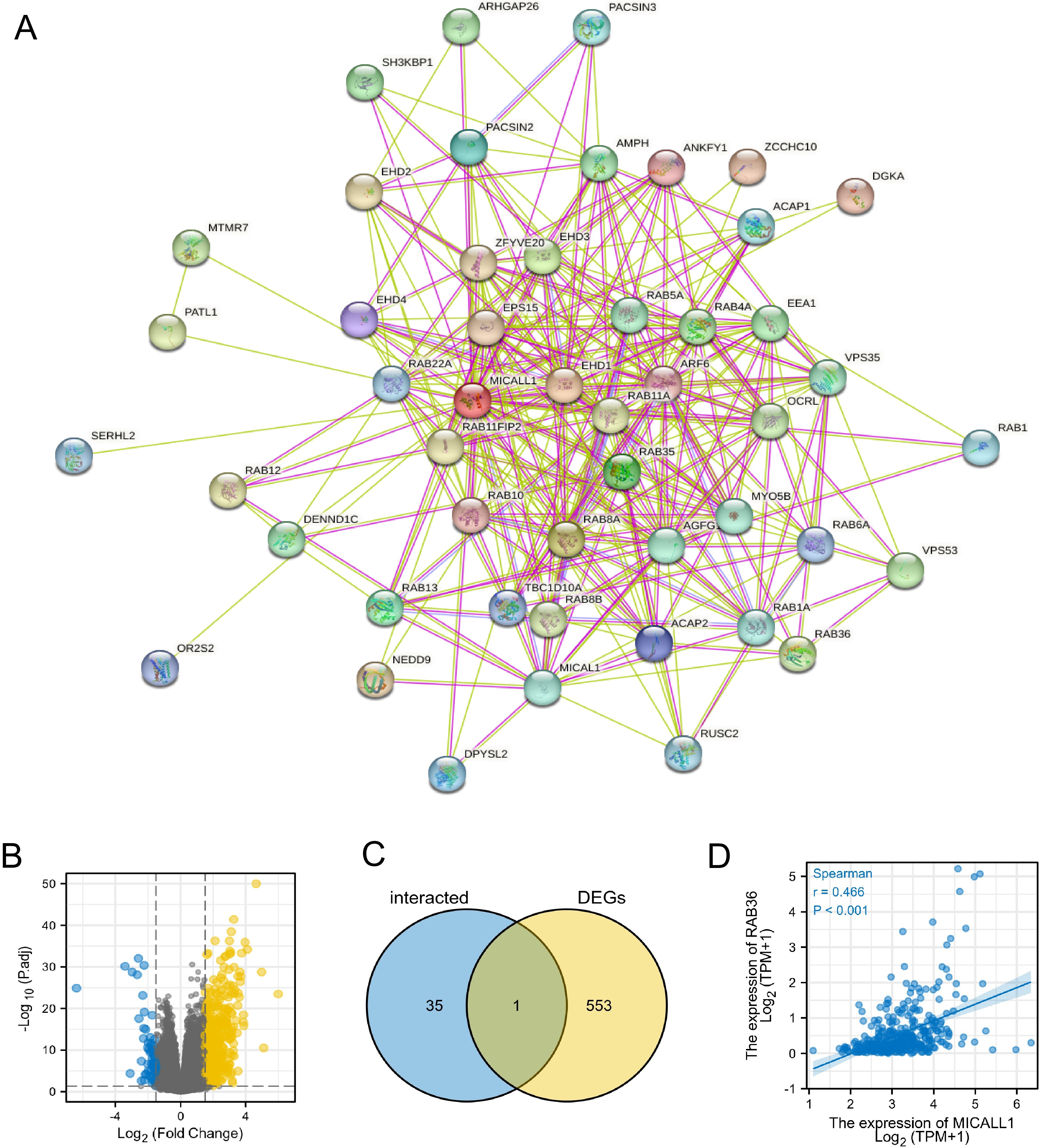
Function enrichment analysis of MICALL1 in LIHC using Gene Ontology (GO), KEGG, and GSEA. (A&B) Significantly enriched GO annotations (A) and KEGG pathways (B) of MICALL1 in LIHC cohort. (C–H) Enrichment plots from GSEA. (C) Hallmark_KRAS_signaling_DN, (D) Hallmark_inflammatory_Response, (E) Hallmark_apical_junction, (F) Hallmark_KRAS_signaling_UP, (G) Hallmark_epithelial_mesenchymal_transition, (H) Hallmark_TNF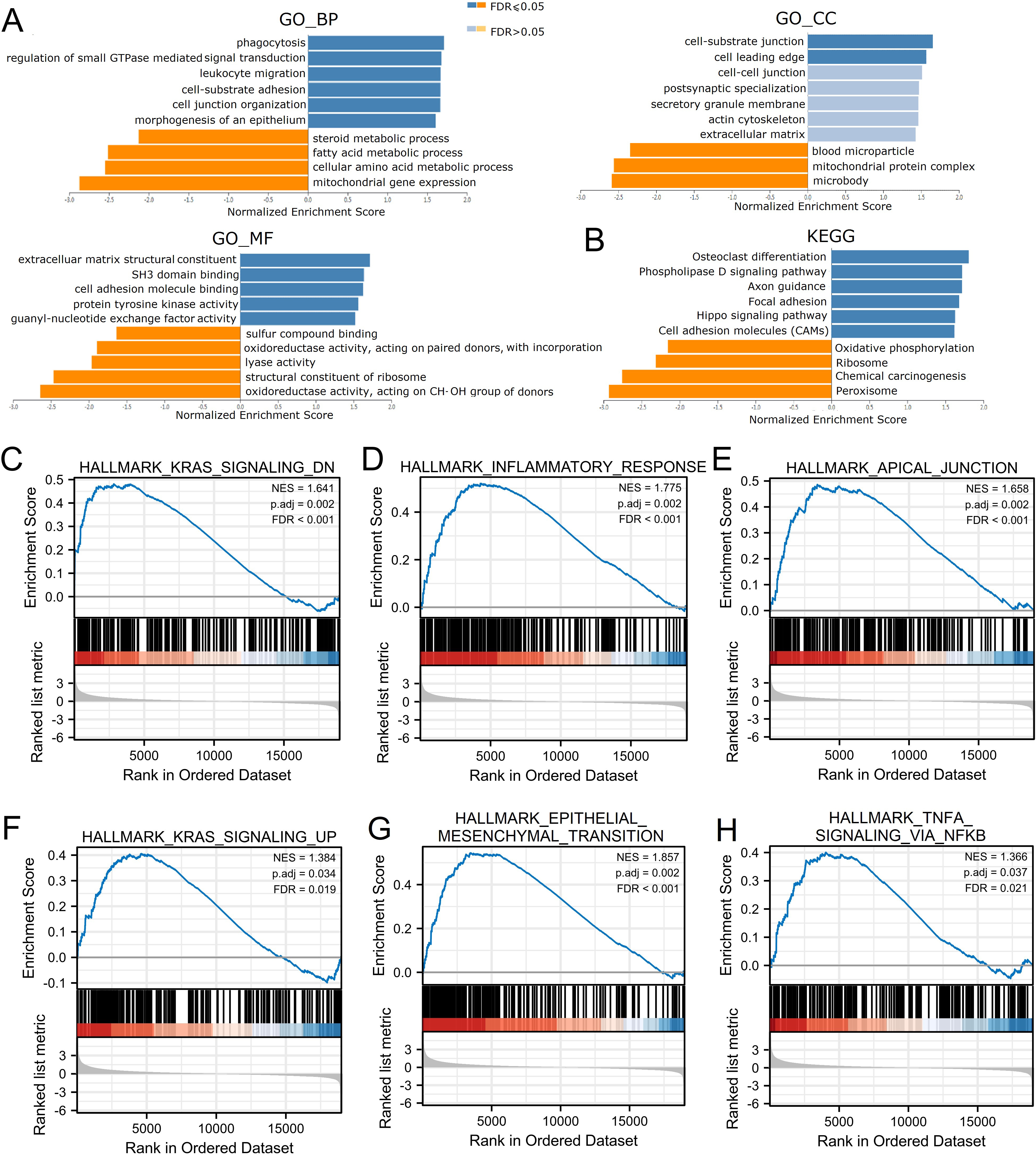
The correlation of MICALL1 and immune infiltration in LIHC. (A) A comparison of immune cells with high or low expression of MICALL1. (B–E) Correlations between MICALL1 expression and immune infiltration levels in LIHC, (B) Th2 cells, (C) NK CD56bright cells, (D) TFH, (E) Macrophages.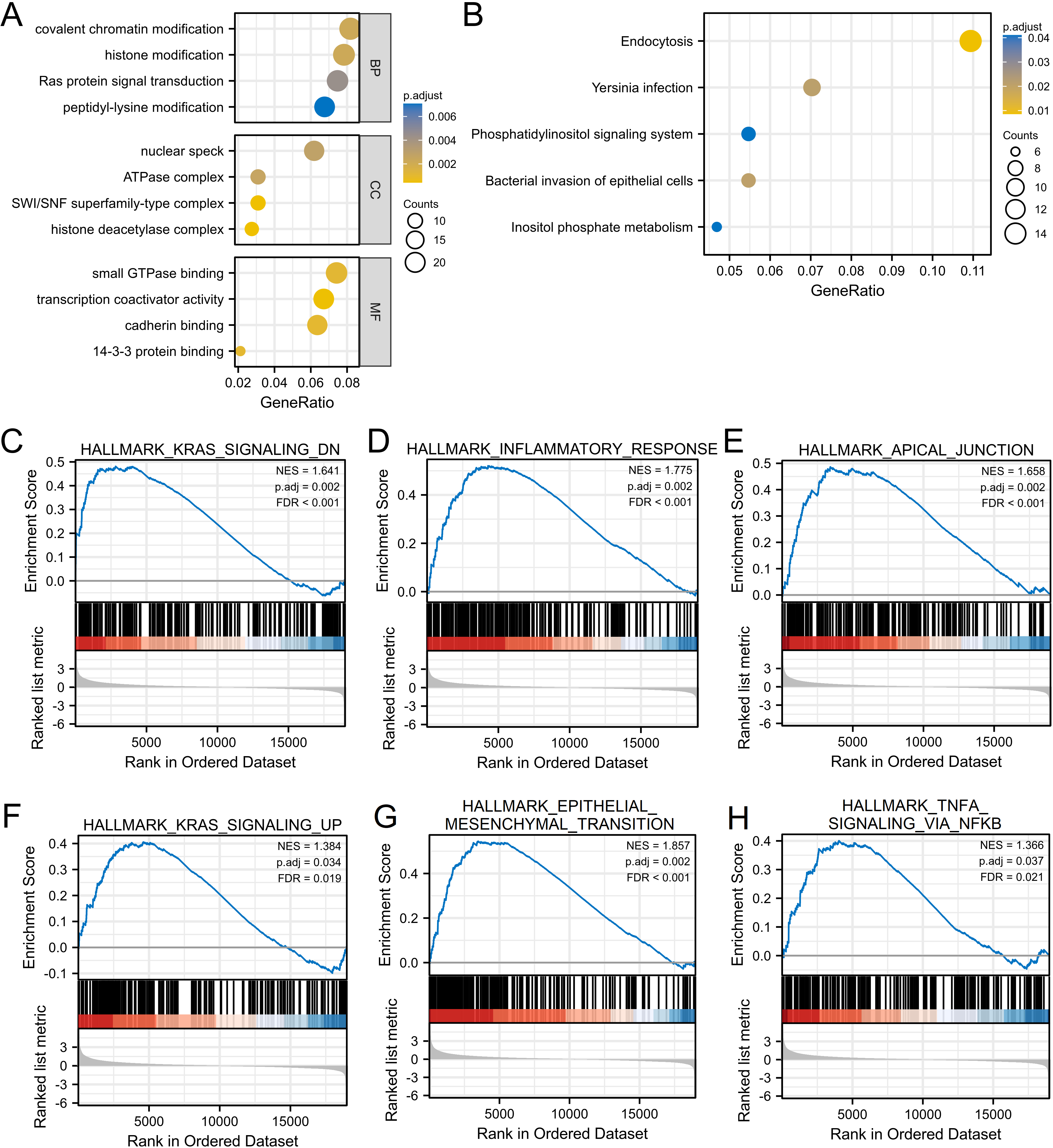
Since LIHC patients with high MICALL1 expression have worse OS, DSS, PFI than those with low MICALL1 expression, we explored the possible cellular mechanism through GO, KEGG and GSEA. As shown in Fig. 5A and B, MICALL1 co-expressed genes participate primarily in regulation of small GTPase mediated signal transduction cell-substrate adhesion and cell junction organization, while the activities like mitochondrial gene expression and multiple metabolic processes were inhibited. KEGG pathway analysis showed enrichment in microRNAs in cancer, focal adhesion, phospholipase D signaling pathway and Hippo signaling pathway, etc. We also found that K-RAS pathway and TNF
The association between MICALL1 and immune infiltration
We explored the association between MICALL1 expression and immune cell infiltration level quantified by ssGSEA in LIHC using Spearman correlation. MICALL1 was positively associated with levels of T helper type 2 (Th2) cells (
Discussion
To the best of our knowledge, the present study was the first one to analyze the associations between MICALL1 expressions and prognosis value of patients with LIHC. The study showed that high expression of MICALL1 were not only found in LIHC tissues, MICALL1 expression was also associated with unfavorable outcomes in LIHC patients. Recent studies have shown that the other member of MICALs, MICALL2, was involved in cancer cell proliferation [41], tumor EMT progression [18], invasive behavior of collective cell migration [42]. Given the functional significance of MICALL2, several studies suggested that MICALL2 might be a reliable marker for predicting prognoses of colon adenocarcinoma as well as kidney renal clear cell carcinoma patients [43, 44]. Since MICALL1 and MICALL2 share similar sturctures, both of which have CH, LIM and CC domains [14], we hypothesized that MICALL1 might also involved in LIHC malignant behavior. Through the analyses of TCGA data, as well as data from UALCAN group, we noticed that LIHC has higher MICALL1 mRNA and protein levels, indicating that MICALL1 would be beneficial in promoting LIHC progression.
MICALL1 is involved in several cellular processes, including plasma membrane tubulation, protein localization to endosome and slow endocytic recycling. MICALL1 exists in a closed conformation, it could change to an opened due to interaction of Rab. It has been well established that Rab35 and MICALL1 promote the recruitment of Rab8, Rab13 and Rab36 to Arf6-positive recycling endosomes during neurite outgrowth [45]. Furthermore, MICALL1/Rab13 protein complex regulated EGFR trafficking at late endocytic pathways [21]. According to our study, MICALL1 is closely related to Rab36. Rab36 is one of the less-known Rab family member and is suspected to be involved in vesicular traffic, which is similar with the function of MICALL1. Rab36 shows prognostic ability in low-grade glioma [46], and is reported to promote bladder cancer cell growth and invasion [47]. To date, few studies have examined the role of MICALL1 in the tumor. Through survival analyses by Kaplan-Meier analysis we determined that patients with higher MICALL1 mRNA expression levels had shorter OS, DSS and DFI in patients, while multivariate Cox analysis confirmed that high MICALL1 expression was an independent risk factor for OS and DSS in patients with LIHC. ROC analysis also confirmed the diagnostic value of MICALL1. In addition, GSEA analysis showed that MICALL1 was related with epithelial-mesenchymal transition process. These above results provided evidences that MICALL1 could used as a prognostic and diagnostic biomarker for LIHC.
The mechanism by which MICALL1 takes part in LIHC progression is not yet known. As noted earlier, MICALL1 contains a unique NPF motif that is lacking by other MICALs, with which it binds to EHD1 [48]. Concordant TGF
Hepatocellular carcinoma is the predominant type of primary liver cancers that mostly induced by chronic inflammation, followed by a sequential progression from chronic liver injury to hepatocellular necrosis and regeneration [53]. Meanwhile, many molecular pathways are involved in LIHC carcinogenesis, including Wnt/
Accumulating evidence has shown that immune cell infiltration plays critical roles in the progression of LIHC [59, 60]. This study demonstrated that MICALL1 expression in HCC was positively associated with multiple types of immune cell infiltration, for example, macrophages. Macrophages are an important part of the immune microenvironment in liver [61]. Tumor-associated macrophages (TAM) usually divided into M1 and M2 type. In the early stage of tumor development, TAMs mainly belongs to M1 phenotype and inhibit tumor growth. As tumor progress to malignancy, TAMs gradually transform to M2 type and promotes immunosuppression [61, 62]. So, the progression of LIHC might influenced by dynamic changes in macrophage phenotypes. In addition, we also found that MICALL1 is positively correlated with the modulation of T helper cells, including Th2 and TFH cells. These associations could be indicative of a potent mechanism where MICALL1 modulates T helper cell functions in LIHC. Together, these results demonstrate that the MICALL1 may participate in the progression of LIHC by regulating the infiltration of immune cells.
In conclusion, our results indicated that MICALL1 was a reliable prognostic biomarker for LIHC, and could be a potential target in the development of anti-LIHC therapy. Obviously, this study was only a bioinformatics analysis study, the data were collected from the online platform databases. Further detailed in vitro and in vivo experiments should be done to validate the results and to reveal the detailed mechanisms underline the effect of MICALL1 on LIHC.
Ethics approval and consent to participate
All data used in the study is publicly available, hence no ethical approval was needed.
Consent for publication
Not applicable.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article.
Competing interests
The authors declare that they have no competing interests.
Funding
None.
Authors’ contributions
Conception: JD.
Interrpretation or analysis of data: YY, WZ, YW.
Manuscript: YY and JD.
Revision for important intellectual content: YY and JD.
Supervision: JD.
Footnotes
Acknowledgments
Not applicable.