
Abstract
BACKGROUND:
Hepatocellular carcinoma (HCC) is one of the most serious malignant tumors with a poor prognosis worldwide. Cuproptosis is a novel copper-dependent cell death form, involving mitochondrial respiration and lipoylated components of the tricarboxylic acid (TCA) cycle. Long non-coding RNAs (lncRNAs) have been demonstrated to affect the tumorigenesis, growth, and metastasis of HCC.
OBJECTIVE:
We explored the potential roles of cuproptosis-related lncRNAs in predicting the prognosis for HCC.
METHODS:
The RNA-seq transcriptome data, mutation data, and clinical information data of HCC patients were downloaded from The Cancer Genome Atlas (TCGA) database. The least absolute shrinkage and selection operator (LASSO) algorithm and Cox regression analyses were performed to identify a prognostic cuproptosis-related lncRNA signature. The receiver operating characteristic (ROC) analysis was used to evaluate the predictive value of the lncRNA signature for HCC. The enrichment pathways, immune functions, immune cell infiltration, tumor mutation burden, and drug sensitivity were also analyzed.
RESULTS:
We constructed a prognostic model consisting of 8 cuproptosis-related lncRNAs for HCC. The patients were divided into high-risk group and low-risk group according to the riskscore calculated using the model. Kaplan-Meier analysis revealed that the high-risk lncRNA signature was correlated with poor overall survival [hazard ratio (HR)
CONCLUSIONS:
The novel cuproptosis-related lncRNA signature could be used to predict prognosis and evaluate the effect of chemotherapy for HCC.
Introduction
Hepatocellular carcinoma (HCC), accounting for
Long noncoding RNAs (lncRNAs), a member of non-coding RNAs (ncRNAs) with longer than 200 nucleotides in length, were transcribed by RNA polymerase II, capped, polyadenylated, and edited [9]. lncRNAs might exert their regulatory roles in gene expression through their binding with RNA, DNA, and proteins. Currently, emerging evidence has demonstrated the key roles of many lncRNAs in the tumorigenicity, growth, metastasis, and drug resistance of HCC, such as lncRNA H19, lncRNA HULC, and lncRNA MALAT1 [9, 10]. Therefore, lncRNAs might be potential diagnostic and therapeutic targets in HCC.
Copper is an essential endogenous metal associated with various biological functions, such as mitochondrial respiration, iron metabolism, and oxygen radical detoxification [11]. The regulation of copper concentrations in the cytoplasm is based on the redox signal generated and transduced by mitochondria [12]. Recently, Tsvetkov et al. uncovered a new form of nonapoptotic cell death pathway, copper-dependent cell death (namely cuproptosis) [13]. The new proposed cuproptosis was different from apoptosis, ferroptosis, or necroptosis, which was triggered by targeting copper to mitochondria [13]. The mechanism of cuproptosis depended on the accumulation of copper, active mitochondrial respiration, and the participation of pivotal components of the tricarboxylic acid (TCA) cycle [13]. The discovery of cuproptosis raised new attention on copper level homeostasis in mitochondria and its potential roles in the development and progression of cancers. Previous studies have shown that the altered metabolizing activities of the TCA cycle were an important feature in HCC [14, 15]. In addition, copper could improve the anti-metastatic activity of disulfiram (DSF) in HCC. DSF/copper suppressed the metastasis and epithelial–mesenchymal transition (EMT) of HCC through NF-
In this study, we constructed a prognostic model for HCC based on 8 differentially expressed cuproptosis-related lncRNAs. The model showed better prognostic value for HCC patients compared with other clinicopathological features. Then a nomogram incorporated the cuproptosis-related lncRNA signature and clinicopathological features to predict the survival rate of HCC patients. We also explored the potential immune functions of the lncRNA signature to elaborate on its potential therapeutic value.
Materials and methods
Data collection
The clinical characteristics of HCC patients in the TCGA dataset
The clinical characteristics of HCC patients in the TCGA dataset
HCC, Hepatocellular carcinoma; TCGA, The Cancer Genome Atlas.
RNA-sequence transcriptome data of 424 samples (374 tumor tissues and 50 normal tissues) was downloaded from the TCGA database (
To identify cuproptosis-related lncRNAs, 19 cuproptosis-related genes were retrieved from the previous literature conducted by Tsvetkov et al. Pearson’s correlation analysis was used to assess the relationships between the cuproptosis-related lncRNAs and HCC calculated by R software with the “limma” package. The threshold values
Construction of the cuproptosis-related lncRNA-based prognostic model
The least absolute shrinkage and selection operator (LASSO) Cox regression analysis was used to screen a multigene signature of the cuproptosis-related lncRNAs by the R software with the “glmnet” package. For the training group, the lncRNA-based prognostic risk score for each patient sample was calculated by the following formula with the expression level multiplied regression model (
Predictive nomogram
We construct a prognostic nomogram to predict the 1-, 3-, and 5-year OS of HCC patients. All independent prognostic factors and the lncRNA-based prognostic signature were included in the construction of the prognostic nomogram using the R software with the “regplot” package. The calibration curve, concordance index (C-index), and ROC curve were used to assess the accuracy of the model.
Functional enrichment analysis
The R software with the “clusterProfler” package was used to perform Gene Ontology (GO) annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of cuproptosis-related differentially expressed genes (DEGs). Gene set enrichment analysis (GSEA) was performed to identify the potential functional pathways between the high-risk group and low-risk group according to the cuproptosis-related lncRNA signature. Enriched biological processes and pathways with
Immune-related function analysis
We further compared the 13 immune-related functions in the high-risk group and low-risk group by calculating each HCC sample gene set enrichment analysis (ssGSEA) scores using the R software with the “gsva” package. Besides, CIBERSORT was used to analyze the differences of 22 types of infiltrating immune cells between high-risk group and low-risk group (
Analysis of tumor mutation burden, checkpoint, and drug sensitivity
Tumor mutation burden (TMB) analysis was performed to explore the gene mutation profile between the high-risk and low-risk groups using the R software with the “maftools” package. The potential immune checkpoints, including CTLA-4, PD-1, and PD-L1 were conducted using the R software with the “limma” package between the two risk groups. According to Genomics of Drug Sensitivity in Cancer (GDSC) 2016 (
Cell culture and quantitative real-time PCR (qRT-PCR) assay
The used primer sequences for qRT-PCR
The used primer sequences for qRT-PCR
The human normal liver cell (LC) was a gift from the Medical School of Ningbo University, Ningbo, China, which was derived from stem cells isolated from a healthy human liver. Two human malignant HCC cell lines, HepG2 and Huh-7, were cultured in high-glucose Dulbecco’s Modified Eagle Medium (DMEM, Mediatech lnc., VA, USA) comprising 10% fetal bovine serum (FBS, PAN, Seratech, Germany) at the humidified chamber of 37
Data were analyzed using the R software (version 4.0.5;
Results
Identification of cuproptosis-related differentially expressed lncRNAs
We firstly collected the transcriptome data of 420 tissues (370 HCC tissues and 50 normal tissues) from TCGA database. Then, the patients were divided randomly into a training group (185 patient samples) and a test group (185 patient samples). Based on the study conducted by Tsvetkov et al., 19 cuproptosis-related genes were collected simultaneously(Supplementary Table 1) [13]. Subsequently, 994 cuproptosis-related lncRNAs were identified by Pearson’s correlation analysis (
Identification of a prognostic cuproptosis-related lncRNA signature
Identification of cuproptosis-related lncRNA signature with prognostic value for hepatocellular carcinoma (HCC) patients. (A, B) LASSO Cox regression with a 10-fold cross-validation for the prognostic value of the cuproptosis-related 8 lncRNAs. (C) Forest plot of the prognostic values of the cuproptosis-related lncRNA signature using multivariate Cox regression analysis. (D) A heatmap showed the Pearson’s correlations between the 8 lncRNAs and the 19 cuproptosis genes.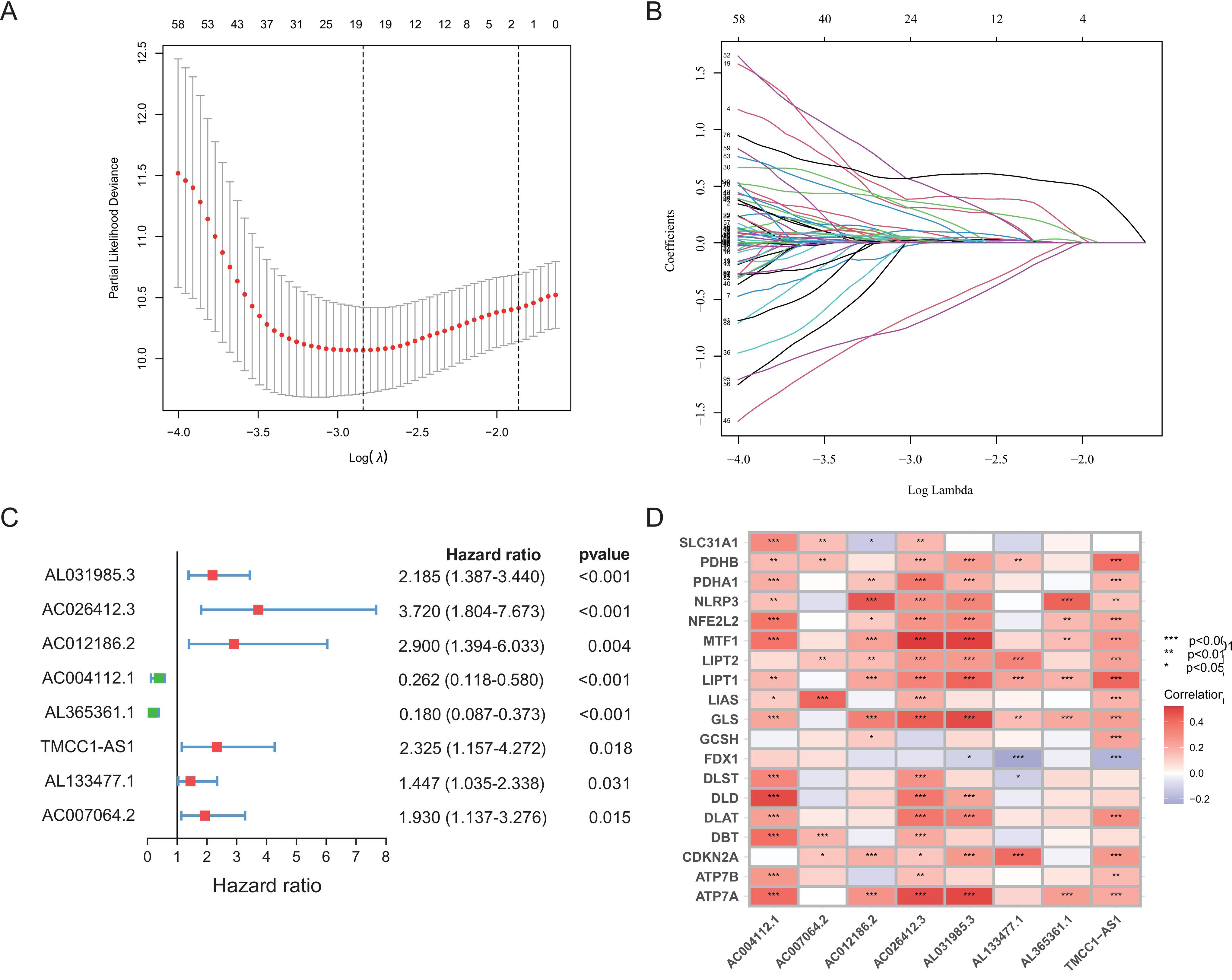
Prognostic risk scores calculated using the cuproptosis-related lncRNA prognostic signature. (A) Risk scores distribution of the high-risk and low-risk patients. (B) Survival status distribution of the high-risk and low-risk patients. (C) Heatmaps of the 8 lncRNAs expressions in the high-risk and low-risk groups. (D) Kaplan–Meier curves for overall survival (OS) of HCC patients in the high-risk and low-risk groups.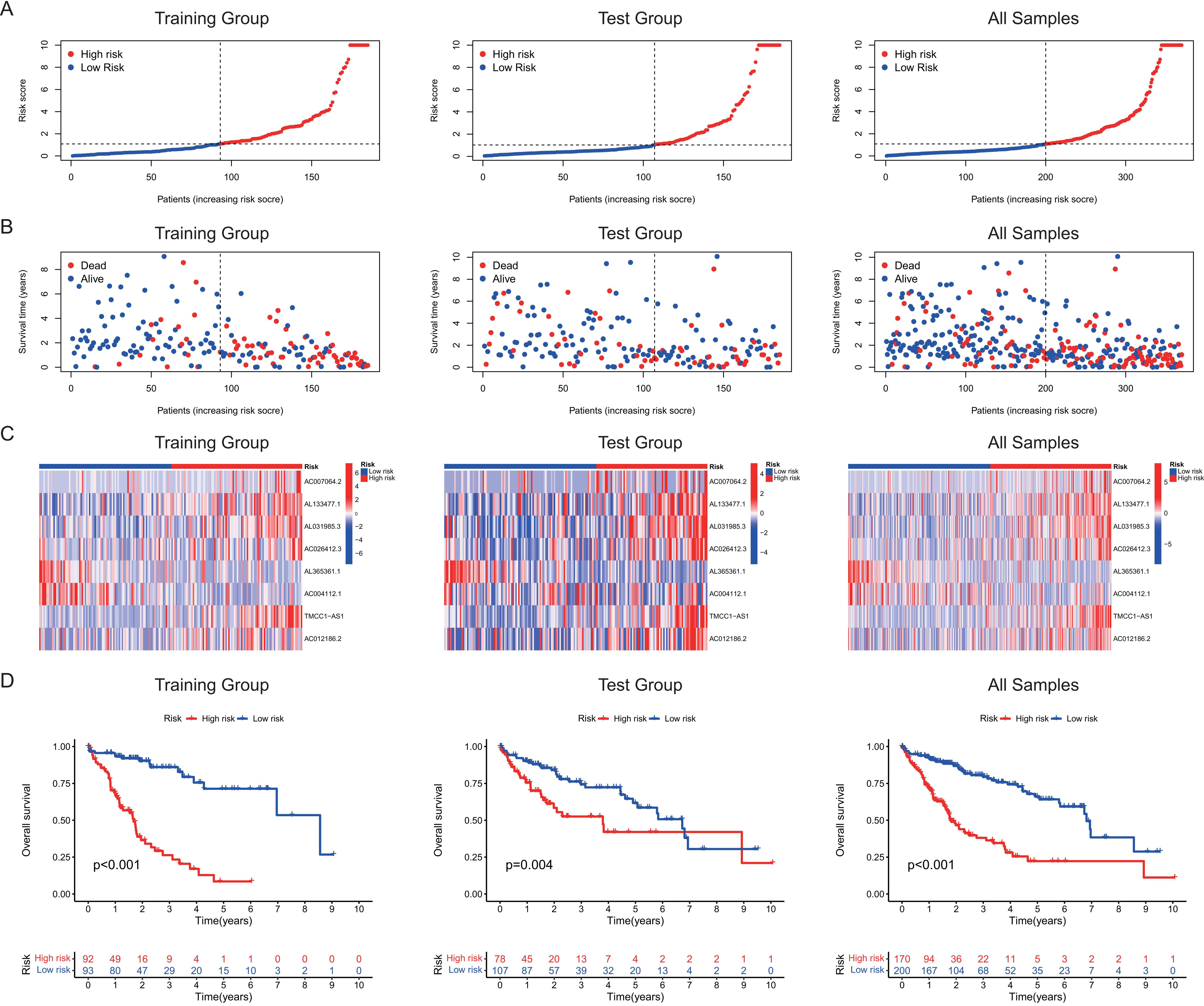
Univariate Cox regression analysis identified 204 significant cuproptosis-related lncRNAs (Supplementary Table 3), which were included in the LASSO algorithm and multivariate Cox regression analysis. Finally, 8 cuproptosis-related lncRNAs (AC004112.1, AC007064.2, AC012186.2, AC026412.3, AL031985.3, AL133477.1, AL365361.1, and TMCC1-AS1) were identified as independent prognosis predictors of HCC, which were used to establish a prognostic cuproptosis-related lncRNA signature (Fig. 1A–C and Supplementary Table 4). Among these 8 lncRNAs, five lncRNAs (AC012186.2, AC026412.3, AL031985.3, AL133477.1, and TMCC1-AS1) were associated with unfavorable prognosis (Supplementary Fig. 1). A heatmap showed the correlations between the 8 lncRNAs and the 19 cuproptosis-related genes (Fig. 1D and Supplementary Table 5). The risk score formula was as follows: (
Identification of the cuproptosis-related lncRNA signature as an independent prognostic factor. Univariate (A) and multivariate Cox regression analyses (B) of the associations between clinicopathological factors (including the lncRNA signature) and OS. (C) Time-dependent ROC curves for OS of the lncRNA signature at 1, 3, and 5 years. Principal component analysis (PCA) of the high-risk and low-risk groups based on the (D) whole-genes, (E) cuproptosis genes, cuproptosis-related lncRNAs, and (F) the risk model (the lncRNA signature). Patients with the high-risk and low-risk scores are indicated in blue and red, respectively. (H) ROC curves for validation of the prognostic value of the cuproptosis-related lncRNA signature. (I) A heatmap for cuproptosis-related lncRNA prognostic signature and clinicopathological features.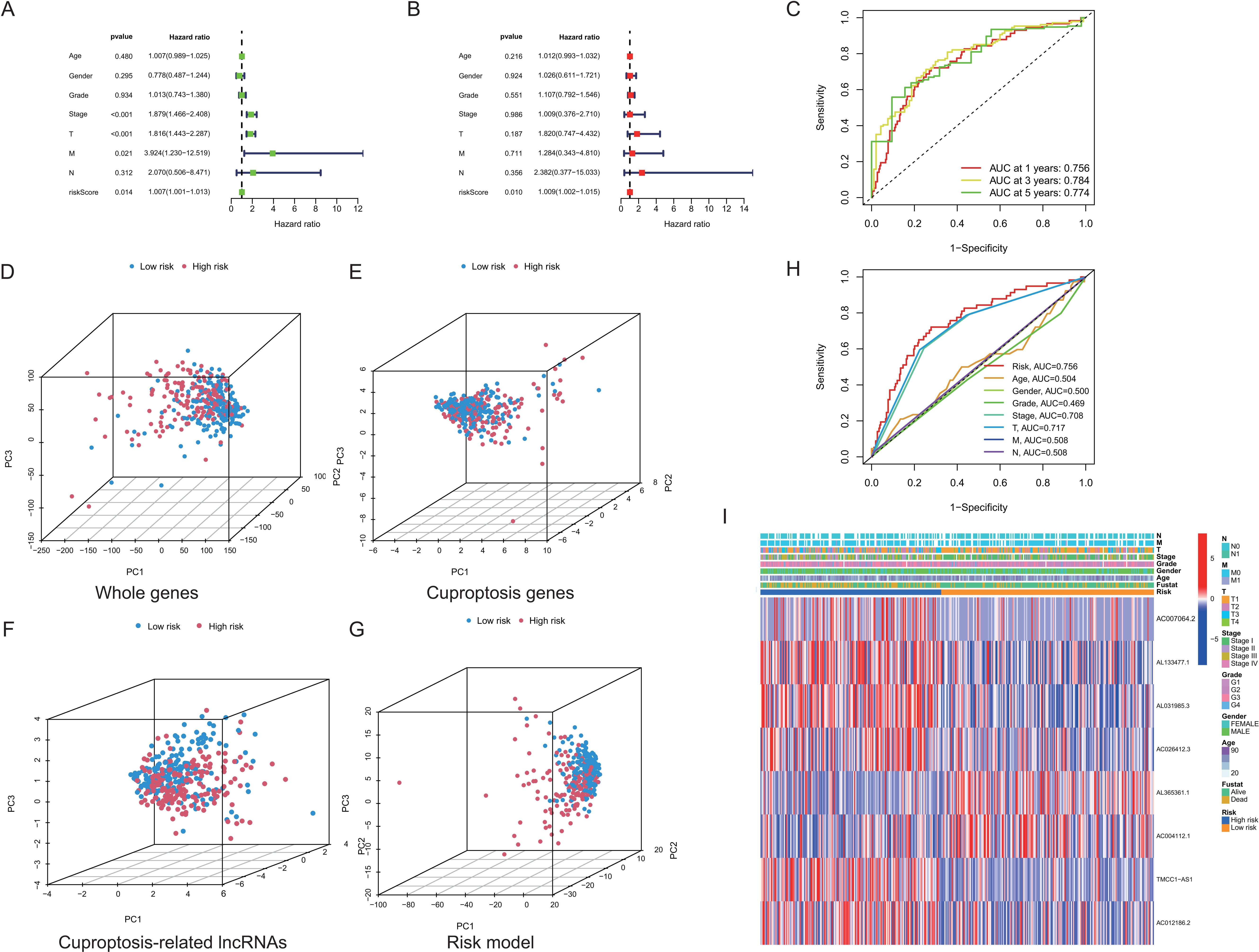
Next, we performed the univariate and multivariate Cox regression analyses to evaluate the independent prognostic factor of the cuproptosis-related lncRNA signature for HCC patients. The results of univariate Cox regression showed that the model [hazard ratio (HR): 1.007, 95% confidence interval (CI): 1.001–1.013;
Kaplan-Meier survival curves analysis in 7 subgroups of HCC patients. (A) Age 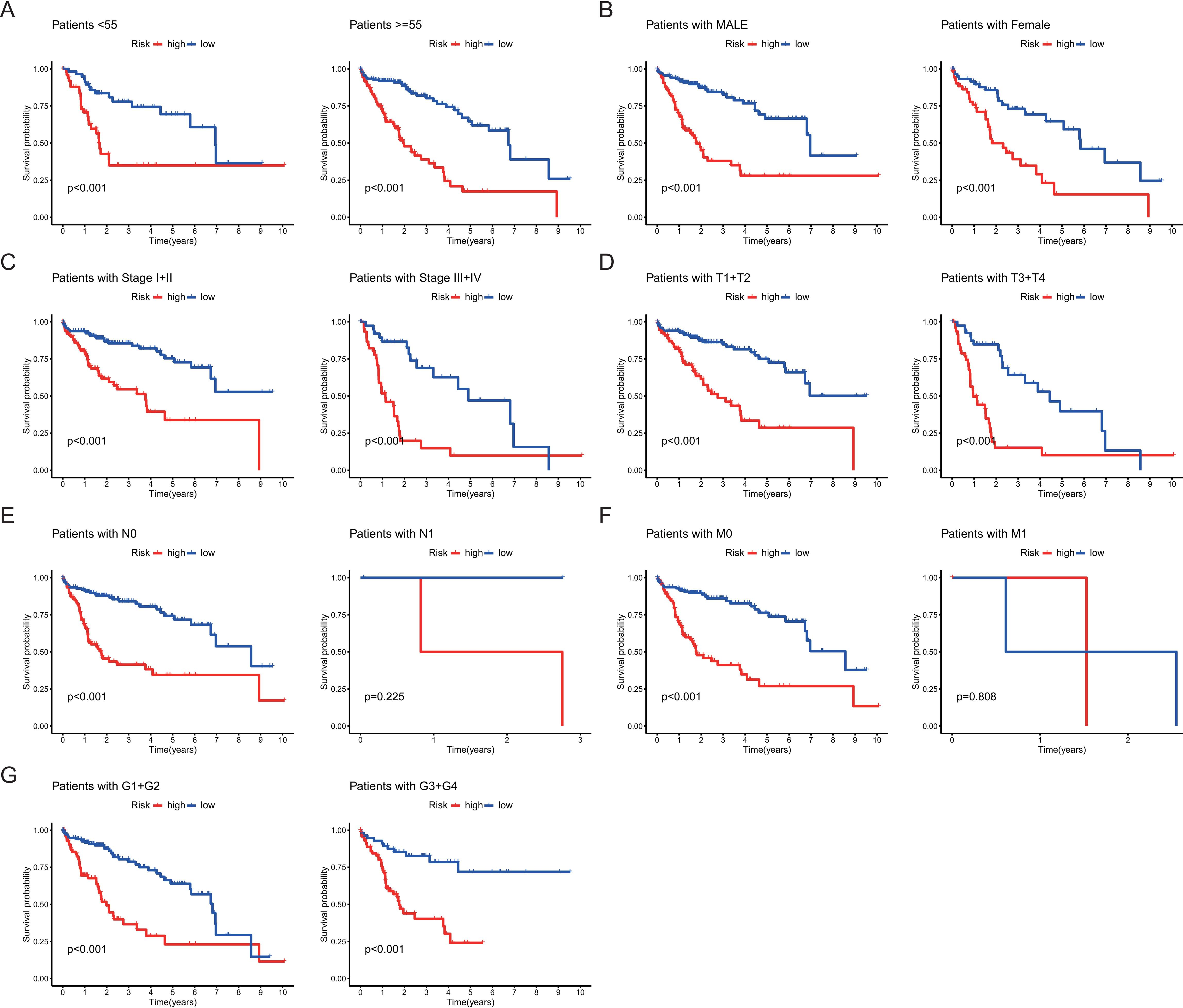
Kaplan-Meier survival curve was performed to evaluate the OS rates stratified by clinicopathological factors between the high-risk group and low-risk group. Results were presented as subgroups stratified by age, gender, clinical stage, grade, T stage, N stage, and M stage (Fig. 4A–G). Although the differences were not observed in the OS of two subgroups (patients with N1 and M1) between the two risk groups, the other 12 clinical groups showed that the patients in the high-risk group had a worse prognosis compared to patients in the low-risk group (Fig. 4A–G).
A nomogram of (OS prediction for HCC patients. (A) A nomogram incorporated the cuproptosis-related lncRNA signature and clinicopathological factors for predicting 1-, 3-, and 5-year survival of HCC patients. (B) Calibration curve of the 1-, 3-, and 5-year survival prediction accuracy for the nomogram. (C) Concordance index (C-index) for the clinical practicality evaluation of the nomogram.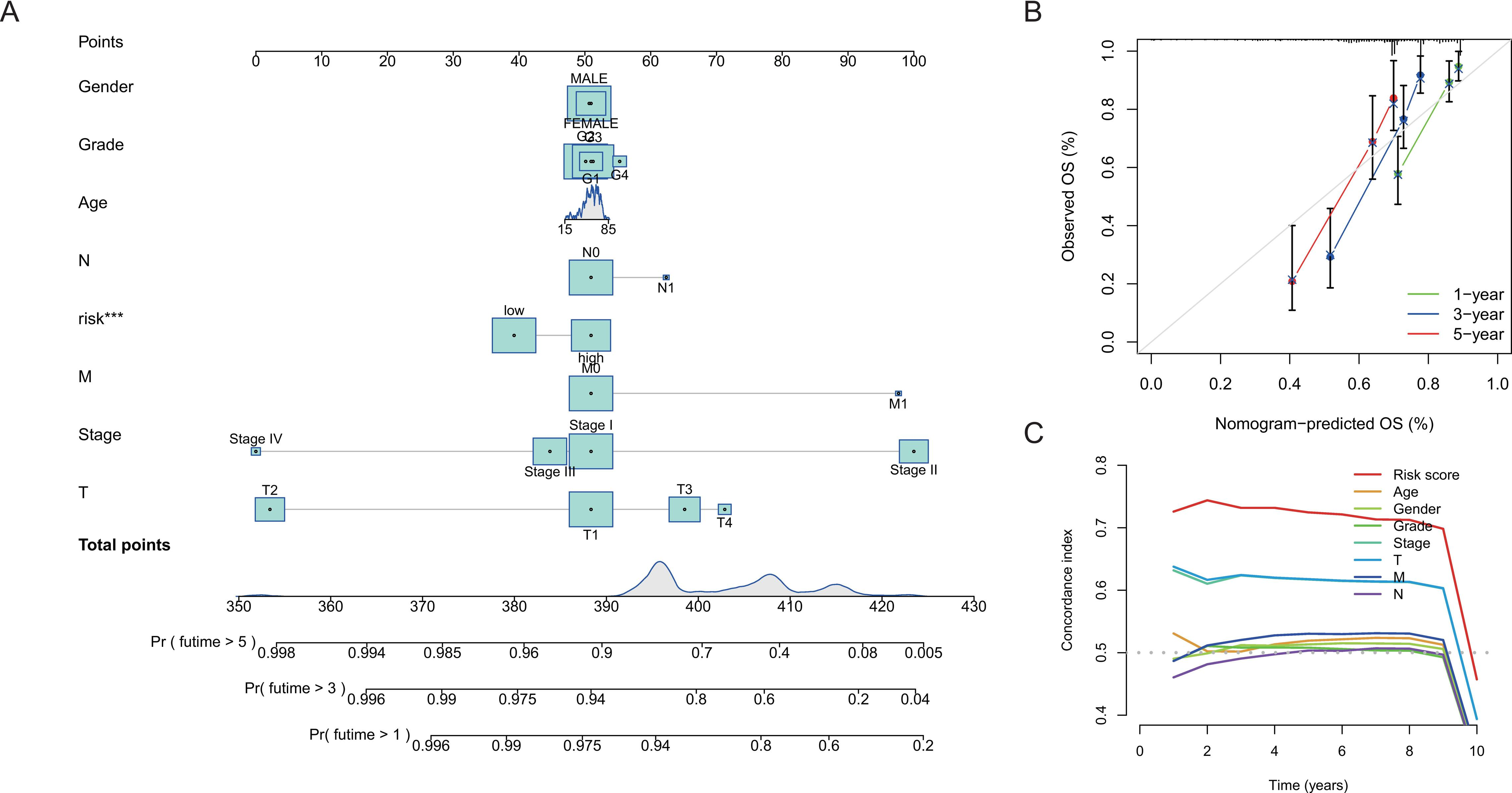
To evaluate the 1-year, 3-year, and 5-year survival probabilities for HCC patients, we constructed a clinically diagnostic nomogram incorporating the cuproptosis-related lncRNA signature and several clinicopathological features (Fig. 5A). The calibration curves of the nomogram for 1-year, 3-year, and 5-year indicated that the nomogram-predicted OS was close to the actual OS rates (Fig. 5B). Besides, the C-index of the risk model was
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses for differentially expressed genes (DEGs) between the high-risk group and the low-risk group. (A) Bubble plots of GO. (B) Bubble plots of KEGG. 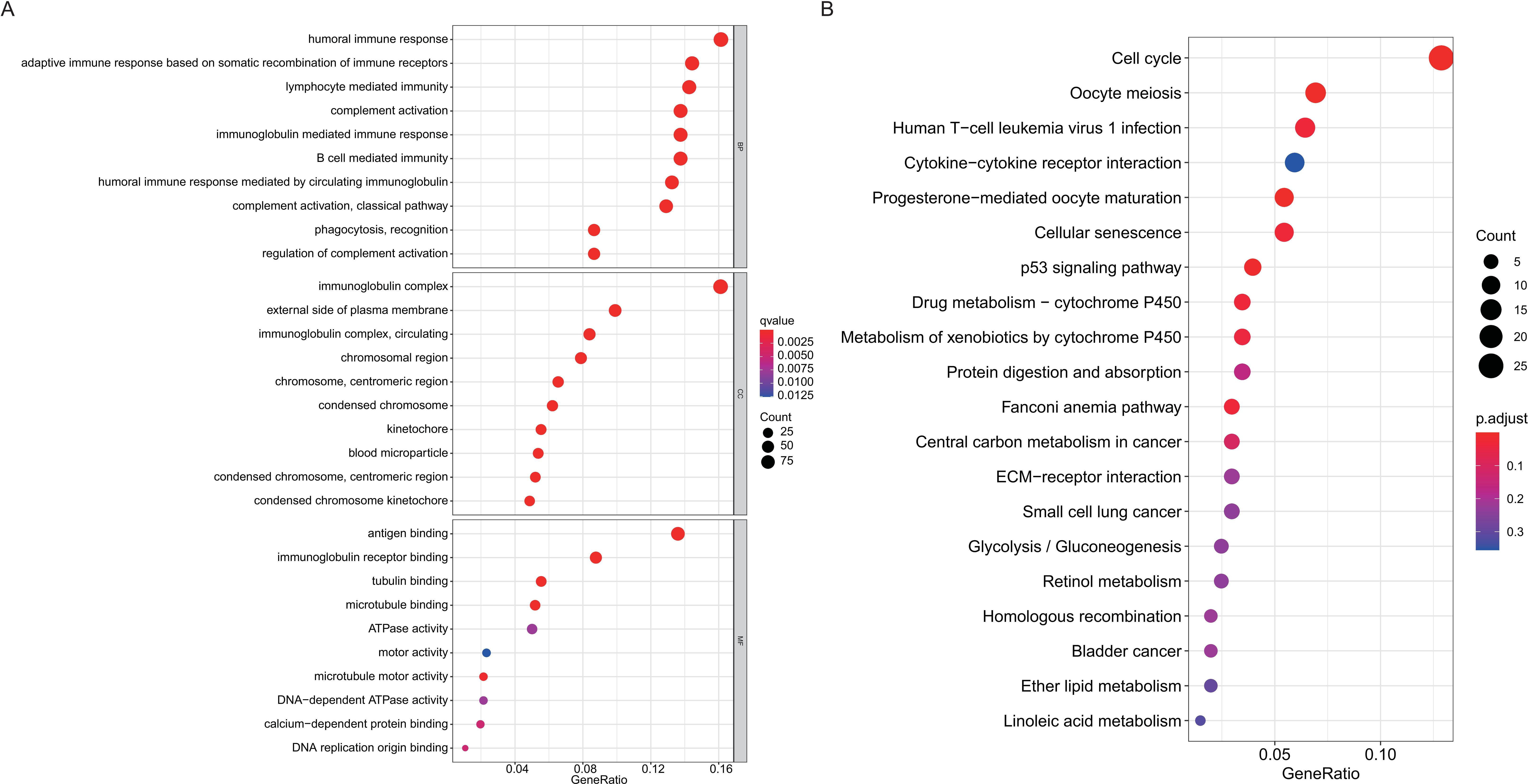
GO enrichment and KEGG pathway analyses were used to explore the functional roles of the DEGs in patients with HCC. GO analysis showed that the DEGs were mostly enriched in humoral immune response, immunoglobulin complex, adaptive immune response, lymphocyte mediated immunity, and antigen binding (Fig. 6A and Supplementary Table 6). KEGG analysis indicated that DEGs were mostly enriched in the cell cycle signaling pathway, oocyte meiosis, human T-cell leukemia virus 1 infection, and cytokine-cytokine receptor interaction (Fig. 6B and Supplementary Table 6).
Immune-related functions analysis of the cuproptosis-related lncRNA signature. (A) A boxplot of the ssGSEA scores of 13 immune-related functions between the high-risk and low-risk groups. (B) A boxplot of CIBERSORT analysis of 22 subtypes of immune infiltration cells between the high-risk and low-risk groups.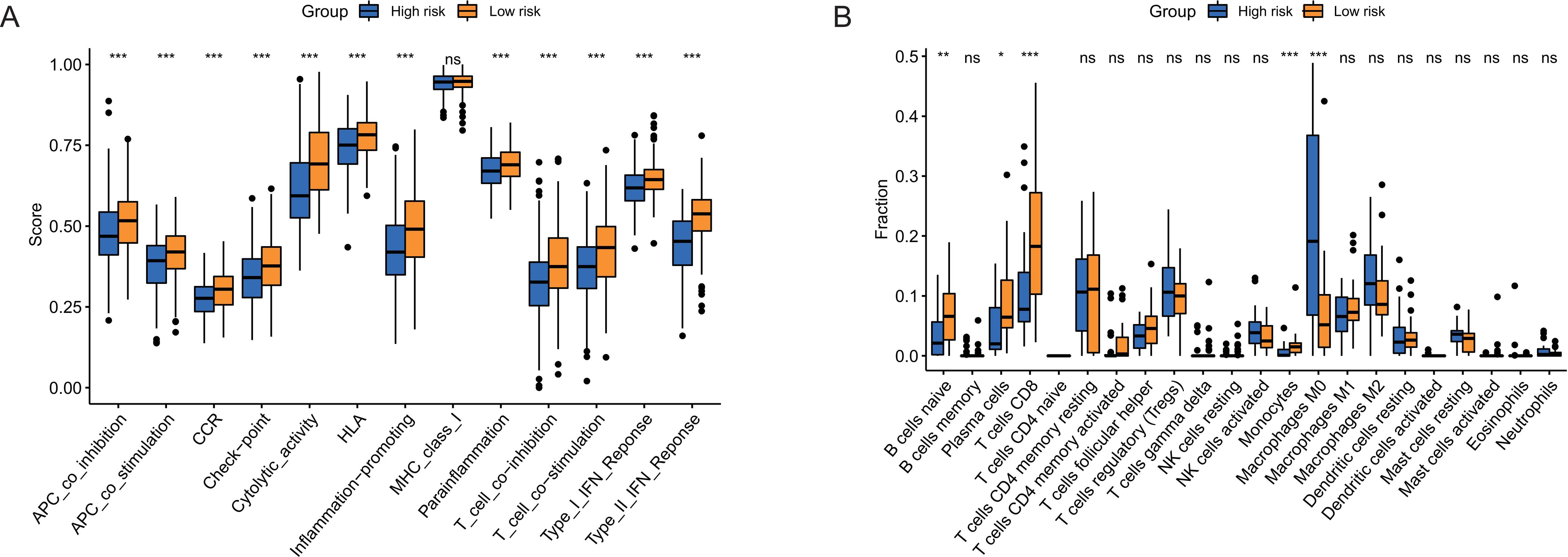
Analysis of tumor mutation burden (TMB), checkpoint, and drug sensitivity in the high-risk and low-risk groups. (A–B) Waterfall plot of 20 mutated genes in the high-risk and low-risk groups. (C) The TMB level in the high-risk and low-risk groups. (D) The expression level of potential immune checkpoints in the high-risk and low-risk groups. (E) The expression levels of TNFRSF4, TNFSF4, PDCD1LG2, and TNFRSF14 in HCC cell lines. (F–K) Drug sensitivity of 5-fluorouracil, doxorubicin, gemcitabine, paclitaxel, sorafenib, and sunitinib for HCC treatment in the high-risk and low-risk groups.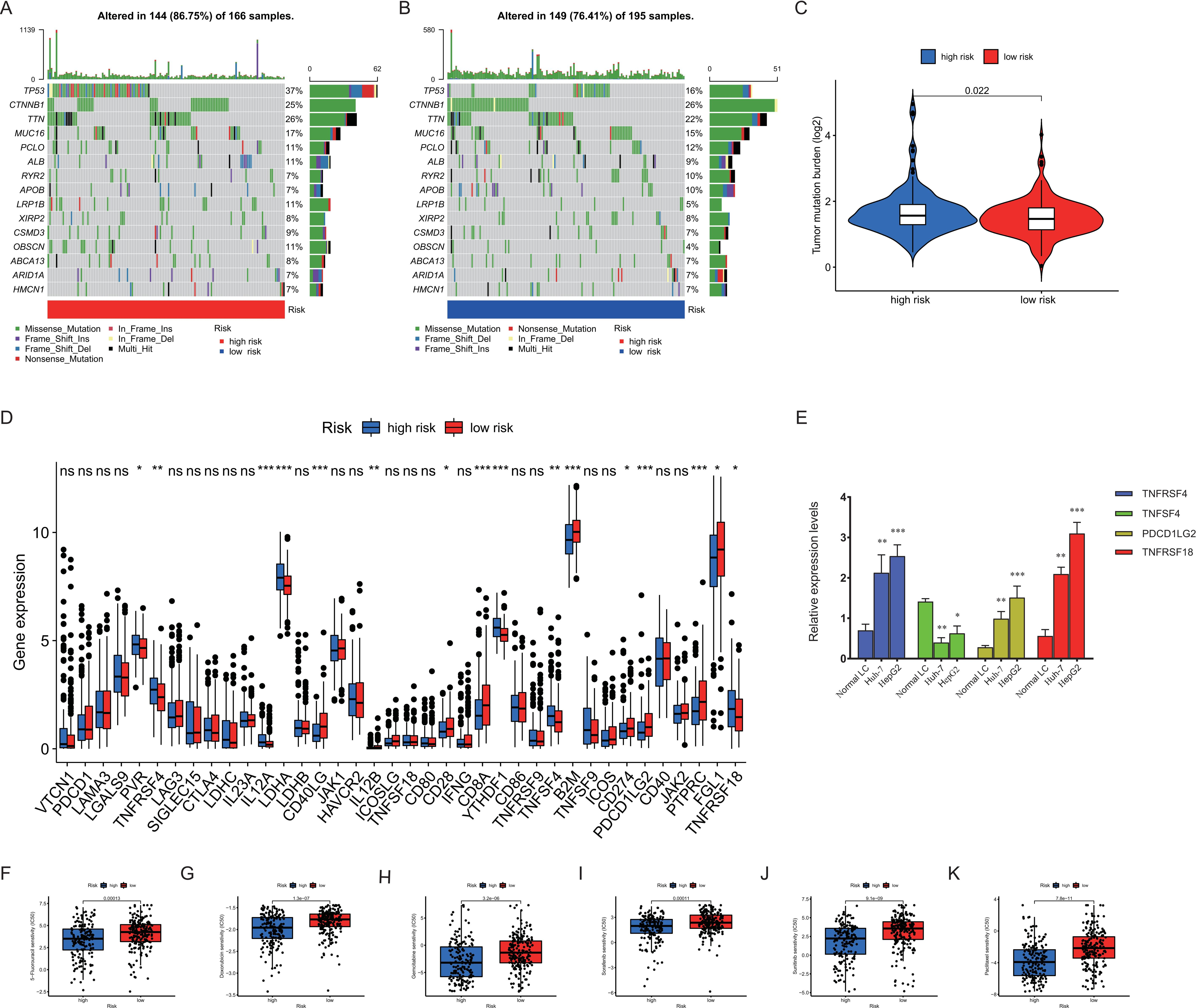
To further evaluate the associations between the risk score and immune function status, we conducted the enrichment scores of immune cell subpopulations and related functions based on ssGSEA. We found that the immune-related functions, including Type interferon (IFN) response, antigen presenting cells (APCs) co-stimulation and co-inhibition, CC chemokine receptor (CCR), T cell co-inhibition and co-inhibition, human leukocyte antigen (HLA), checkpoint, cytolytic activity, and inflammation promoting were all significantly lower in the high-risk group compared with that in the low-risk group (All
Then, we analyzed mutational status and TMB in the high-risk and low-risk groups. The top 20 genes with the highest mutation frequency were presented in a waterfall plot. As shown in Fig. 8A and B, missense mutations were the most common genetic alteration among somatic mutations. TP53, CTNNB1, and TTN were the three genes with the highest mutation frequency in both the high-risk and low-risk groups (Fig. 8A and B). Besides, the TMB between the two risk groups was significantly different (
Discussion
In China, HCC ranks as the fourth highest incidence of all malignant tumors, with approximately 470,000 new cases and 420,000 deaths annually, which accounts for approximately 50% of the total deaths caused by HCC globally [18]. Therefore, HCC raises a great economic and societal burden in China as well as affects patients’ quality of life.
Previous studies have described the lncRNA signatures for HCC prognosis and immune response. Xu et al. constructed a 9 ferroptosis-related lncRNAs (CTD-2033A16.3, CTD-2116N20.1, CTD-2510F5.4, DDX11-AS1, LINC00942, LINC01224, LINC01231, LINC01508, and ZFPM2-AS1) prognostic model for HCC [19]. Besides, an autophagy-related four-lncRNA signature (LUCAT1, AC099850.3, ZFPM2-AS1, and AC009005.1) had been reported to be associated with regulatory mechanisms in HCC prognosis [20]. In the current study, we firstly identified a novel 8 cuproptosis-related lncRNA signature based on the patients with HCC of TCGA dataset. We also demonstrated that the lncRNA signature had a valuable predictive performance for HCC OS with higher AUC (0.756). Overall, 5 lncRNAs (AC004112.1, AC026412.3, AL031985.3, AL365361.1, and TMCC1-AS1) out of the 8 candidate lncRNAs had been reported to be correlated with development and prognosis in various tumors. AC004112.1 was a member of seven-lncRNA signature, which was used to assess prognostic risk for papillary thyroid carcinoma [21]. AC026412.3 and AL031985.3 were two components of a pyroptosis-related lncRNA risk signature related to prognosis and immune infiltration of HCC, suggesting a potential link between pyroptosis and immune regulation in HCC [22]. Besides, AL031985.3 and TMCC1-AS1 had also been integrated into several models correlated with ferroptosis and autophagy to predict the outcomes, immune cell infiltration, and immunotherapy response for HCC patients [23, 24]. AL365361.1 was also reported to be associated with OS in neck squamous cell carcinoma and ovarian cancer [25, 26]. Next, we found that synapse associated protein 1 (SYAP1) might be a target gene of lncRNA AC012186.2 using starbase (
More importantly, we also explored the potential pathways involved in DEGs between the high-risk and low-risk groups. In our study, GO and KEGG enrichment analysis showed that many immune-related biological processes and pathways were enriched, such as humoral immune response, adaptive immune response, lymphocyte mediated immunity, and cell cycle. Therefore, we then explored the feature of immune-related functions and subtypes of infiltrating immune cells between the high-risk group and low-risk group. Results showed that all 13 immune-related functions were significant lower in the high-risk group than that in the low-risk group except for MHC class I. The impaired immune functions in the high-risk group might contribute to HCC carcinogenesis [27]. For example, T cell activation and function were regulated by co-stimulatory and co-inhibitory pathway. The proliferation, differentiation, and survival of T cells required co-stimulatory signals delivered by the APCs, making co-stimulation necessary to induce immune responses [27]. Antitumor immune responses of T cells also depended on efficient presentation of tumor antigens and co-stimulatory signals by host APCs [27]. The weaken co-stimulatory and co-inhibitory of APCs and T cells in the high-risk patients might lead to reduced tumor antigen presentation and killing, thus facilitating the immune escape of HCC. Therefore, co-stimulatory and co-inhibitory markers have been used as therapeutic targets of tumor immunotherapy [27, 28]. Moreover, type II IFN response played important functions on immune surveillance, promoting tumor antigen recognition and elimination in cancers [29]. Evidence indicated that IFN could directly activate tumor suppressor gene p53, which might lead to the apoptosis of several tumor cells [30]. Narkwa et al. reported that AFB1 down-regulated the type I IFN signaling pathway through inhibiting the JAK1, STAT1, and OAS3 to induce occurrence of HCC in HepG2 cells [31]. Therefore, the impaired immune functions in the high-risk group might compromise the roles of innate immune system in supervising and defensing HCC.
In addition, CIBERSORT analysis indicated that the proportion of M0 macrophages was significantly higher in the high-risk group compared to that in the low-risk group. Tumor-associated macrophages (TAMs) were essential members of TIICs in promoting tumor inflammation, tumor initiation, progression, and extracellular matrix remodelings, such as M1 macrophages and M2 macrophages [32]. Different from M1 or M2, M0 macrophages might be another type of TAMs or an incompletely differentiated M2 by transcriptomic profiling analysis in ovarian cancer and glioblastoma [33, 34]. Evidence has indicated that M0 macrophages were positive with poor prognosis in lung cancer [35]. Moreover, it was reported that M0 macrophages were associated with worse OS in HCC, indicating the potential predictive role of M0 macrophages for patients with HCC [36].
In fact, TMB has now been used as an immunotherapy biomarker of cancer therapy. The tumors with the higher TMB were more likely to respond better to immune checkpoint blockade (ICB) agents [37]. Our study showed that TP53 and CTNNB1 were the highest mutated genes in the high-risk group and the low-risk group, respectively. TP53 mutation (12%–48%) was the most common mutation in HCC patients [38]. TP53 inactivation, as well as FGF19 and/or CCND1 amplifications were reported to be associated with chromosomal instability of HCC [39]. Besides, TP53 inactivation might lead to dysregulation of cell-cycle activity, which was correlated with poorly differentiated, vascular invasion, and poor prognosis for HCC [40]. CTTNB, encoding
The strength of this study was that we explored the correlations between cuproptosis-related lncRNAs and the prognosis of HCC based on the TCGA database. The constructed cuproptosis-related 8-lncRNA signature showed a good performance in predicting the prognosis for HCC patients compared with other clinical features. In addition, we also assessed the immune-related functions and pathways related to the cuproptosis-related 8-lncRNA signature, which might provide potential insights for the patient selection for more effective anti-tumor immunotherapies. However, there were several limitations in our study. First, our results were based on a retrospectively single database source. Second, our findings were mainly based on integrative bioinformatics, thus prospective and multicenter clinical studies were needed to verify the prognostic value of our model. Third, further functional experiments were needed to elucidate the biological mechanisms of how cuproptosis-related lncRNAs regulated the process of HCC.
Taken together, we firstly constructed a novel cuproptosis-related lncRNA signature for predicting the prognosis of HCC patients. A predictive nomogram based on the cuproptosis-related lncRNA signature was constructed for HCC. Besides, the cuproptosis-related lncRNA signature might be correlated with immune infiltration levels, especially M0 macrophages, and predict the immunotherapy response of HCC.
Author contributions
For every author, his or her contribution to the manuscript needs to be provided using the following categories:
Conception: Jianshuai Jiang, Yanqing Liu.
Interpretation or analysis of data: Yanqing Liu.
Preparation of the manuscript: Yanqing Liu.
Revision for important intellectual content: Jianshuai Jiang, Yanqing Liu.
Supervision: Jianshuai Jiang.
Ethic statements
This study design followed the local legislation and institutional requirements in accordance with the Declaration of Helsinki. Our research was approved by the Ethical Committee of the Ningbo First Hospital, and written informed consent was waived because the data were obtained from public databases.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-220259.
sj-pdf-1-cbm-10.3233_CBM-220259.pdf - Supplemental material
Supplemental material, sj-pdf-1-cbm-10.3233_CBM-220259.pdf
sj-pdf-2-cbm-10.3233_CBM-220259.pdf - Supplemental material
Supplemental material, sj-pdf-2-cbm-10.3233_CBM-220259.pdf
sj-xls-1-cbm-10.3233_CBM-220259.xls - Supplemental material
Supplemental material, sj-xls-1-cbm-10.3233_CBM-220259.xls
sj-xls-2-cbm-10.3233_CBM-220259.xls - Supplemental material
Supplemental material, sj-xls-2-cbm-10.3233_CBM-220259.xls
sj-xlsx-1-cbm-10.3233_CBM-220259.xlsx - Supplemental material
Supplemental material, sj-xlsx-1-cbm-10.3233_CBM-220259.xlsx
sj-xlsx-2-cbm-10.3233_CBM-220259.xlsx - Supplemental material
Supplemental material, sj-xlsx-2-cbm-10.3233_CBM-220259.xlsx
sj-xlsx-3-cbm-10.3233_CBM-220259.xlsx - Supplemental material
Supplemental material, sj-xlsx-3-cbm-10.3233_CBM-220259.xlsx
sj-xlsx-4-cbm-10.3233_CBM-220259.xlsx - Supplemental material
Supplemental material, sj-xlsx-4-cbm-10.3233_CBM-220259.xlsx
Footnotes
Acknowledgments
This study was supported by the supported by the Medical and Health Science and Technology Projects of Zhejiang Province (No.2022KY308).
Conflict of interest
The authors declare no conflict of interests.