
Abstract
BACKGROUND:
More and more studies have shown that long non-coding RNA (LncRNA) as a competing endogenous RNA (ceRNA) plays an important role in lung cancer. Therefore, we analyzed the RNA expression profiles of 82 lung cancer patients which were all from Gene Expression Omnibus (GEO).
METHODS:
Firstly, we used BLASTN (evalue
RESULTS:
We intersected the genes of above 4 modules with the differential expression genes: 28 LncRNAs (up: 5, down: 23) and 265 mRNAs (up:11, down: 254). Based on these genes, we picked up 6 LncRNAs (CCDC39, FAM182A, SRGAP3-AS2, ADAMTS9-AS2, AC020907.2, SFTA1P), then set and visualized the LncRNA-miRNA-mRNA ceRNA network with 12 miRNAs related to 12 mRNAs. Finally, we performed downstream analysis of 265 mRNAs by Gene Ontology (GO) enrichment analysis, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis and Protein-Protein Interaction (PPI) network.
CONCLUSION:
After analyzing, we think this study provides a new direction for basic and clinical research related to LAD, and is expected to provide new targets for early diagnosis, prognostic evaluation and clinical treatment of lung cancer.
Introduction
Lung cancer is one of the most frequently occurring tumors in the world. Every year, about 2.1 million people are newly diagnosed with lung cancer [1]. According to “Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries” published in A Cancer Journal for Clinicians (CA) by International Agency for Research on Cancer (IARC) reported that in 2018, there were 2.094 million new lung cancer patients and 1.761 million deaths worldwide, accounting for 11.6% and 18.4% of all cancer incidences and deaths, respectively, ranking first in incidence and mortality. Not only that, because of the poor prognosis of lung cancer and the low five-year survival rate, it has become the number one cause of death for men in more than half of the world’s countries. Especially in China, the incidence and mortality rate are the highest in the population [2]. On the basis of histology, lung cancer is classified into two main subtypes: small cell lung carcinoma (SCLC) and non-small-cell lung carcinoma (NSCLC). Furtherly, NSCLC is divided into three types: lung squamous-cell carcinoma (LUSC), lung adenocarcinoma (LAD), and lung large-cell carcinoma. LAD is the most common type of lung cancer, which makes up approximately 40% of all lung cancer [3].
LncRNAs are a class of non-coding RNA strands larger than 200 nucleotides. As a research object of molecular biology in recent years, it has been shown to be closely related to the occurrence of many diseases, especially tumors [4]. For example, MALAT1 was first discovered in non-small cell lung cancer (NSCLC), and its expression is related to the susceptibility to NSCLC. The prognosis and metastasis closely related [5]. Its mechanism of action is diverse. It can play a role by regulating the expression of genes before transcription, affecting the activity of RNA polymerase through specific transcription factors, regulating the process of RNA splicing, or acting on the epigenetic level [6].
Competing endogenous RNA (ceRNA) has been denied as some transcripts which are able to regulate each other by competing for shared miRNAs at post-transcription level [7]. By finding and analyzing the ceRNAs that competitively combined with miRNAs to change miRNAs’ expression, ceRNA network can link the different functions of protein-coding mRNAs and non-coding RNAs. Because of this character, recently, scientists would like to build the ceRNA network to find the possible pathways which may have important effects on the onset, development and treatment of disease, such as lumbar intervertebral disc degeneration [8], colon adenocarcinoma [9], mesenchymal glioblastoma [10] and so on.
The GEO database is an international public functional gene expression repository created and maintained by NCBI. It has a powerful collection and storage function [11]. It provides a large amount of disease-related gene expression profile information, and high-throughput gene chip. It greatly facilitates the research of diseases, especially the molecular level of tumors. This study based on data from cancer patients in the GEO database analyzed several LncRNAs expression and its possible mechanism of action.
Materials and methods
Data sets
We obtained the LncRNAs gene expression data related to LAD from the GEO database and used these in our study. In order to evaluate LncRNAs’ expression characteristics, we performed sample screening according to the following three criteria: (1) Study of LncRNAs about LAD. (2) The samples include tumor tissues and normal tissues. (3) The probe ID and sequence must be in the platform file to which the sample belongs. Finally, we downloaded the expression matrices GSE85716 (
Reannotation of data
We downloaded the BLAST [12] (Version 2.10.1) analysis tool from NCBI, and used the BLASTN (evalue
Integration of data
Firstly, we used limma packages [15] in R to perform in- group correction on three data sets. Then, all the samples of the three data sets were integrated, and the number of samples was expanded to 82, including 41 tumor samples and 41 normal samples. Finally, in order to avoid a reduction in the reliability of the analysis, we used the sva packages [16] in R to batch normalize the merged data set.
Differential expression analysis
We used the limma package of R to compare tumor tissues with normal tissues for gene difference analysis (|LogFC|
Weighted correlation network analysis
Weighted correlation network analysis (WGCNA) can find modules of highly correlated genes, to relating modules to external sample traits [17]. In our research, mRNAs and lncRNAs between the tumor tissues and normal tissues were analyzed by the WGCNA R software package to get modules which were most related with LAD. In scale free topology model fit, we chose R
LncRNA-miRNA-mRNA ceRNA network
We predicted LncRNA-miRNA interactions for LncRNAs with overlapped differential expression by miRcode [19] (
GO enrichment analysis
GO (
KEGG pathway enrichment analysis
KEGG (
Protein-protein interaction network
We constructed PPI network for 265 mRNA in overlapped differential gene expression by using STRING (
Results
Data collation
Firstly, we downloaded gene expression data and platform information of the GEO database GSE85716 (
Details of lung adenocarcinoma studies data sets from GEO database
Details of lung adenocarcinoma studies data sets from GEO database
Normalization of LncRNAs and mRNA data sets. (a) LncRNA. (b) mRNA. Red represents before normalization and green represents data after.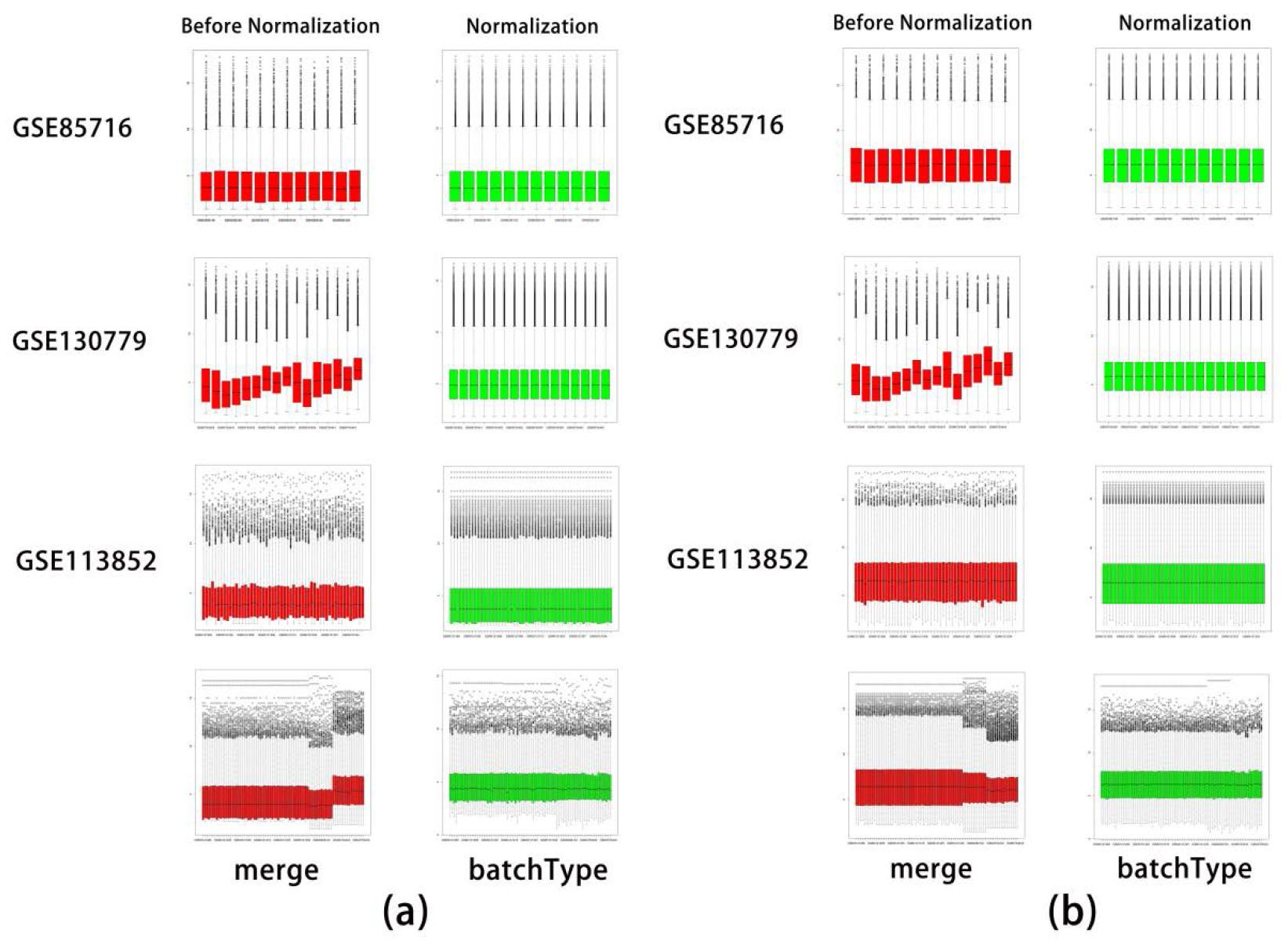
Differential gene expression. The red color means the up-regulated genes; the green color means the down-regulated genes; the black color means no change in genes.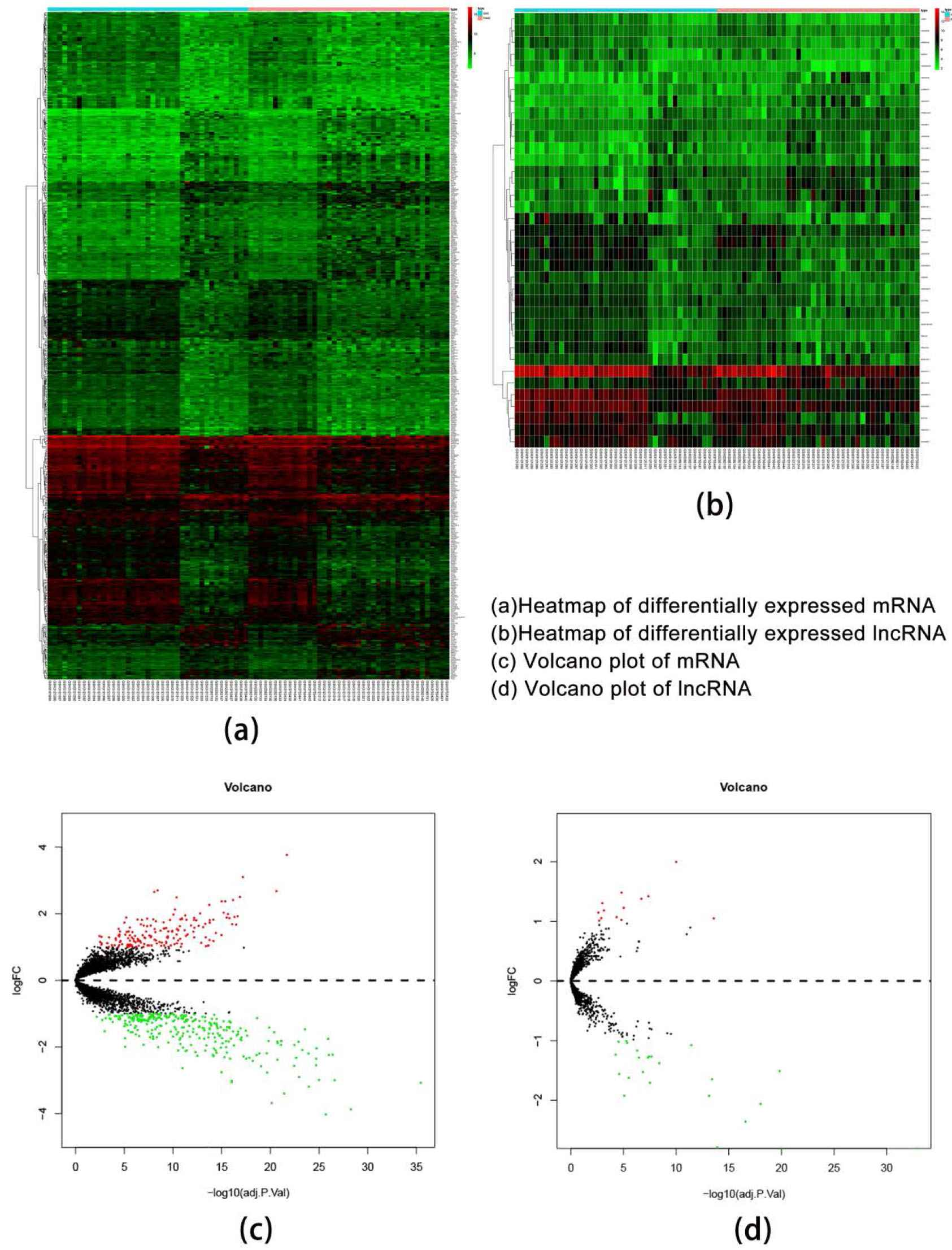
WGCNA of LncRNAs and mRNA.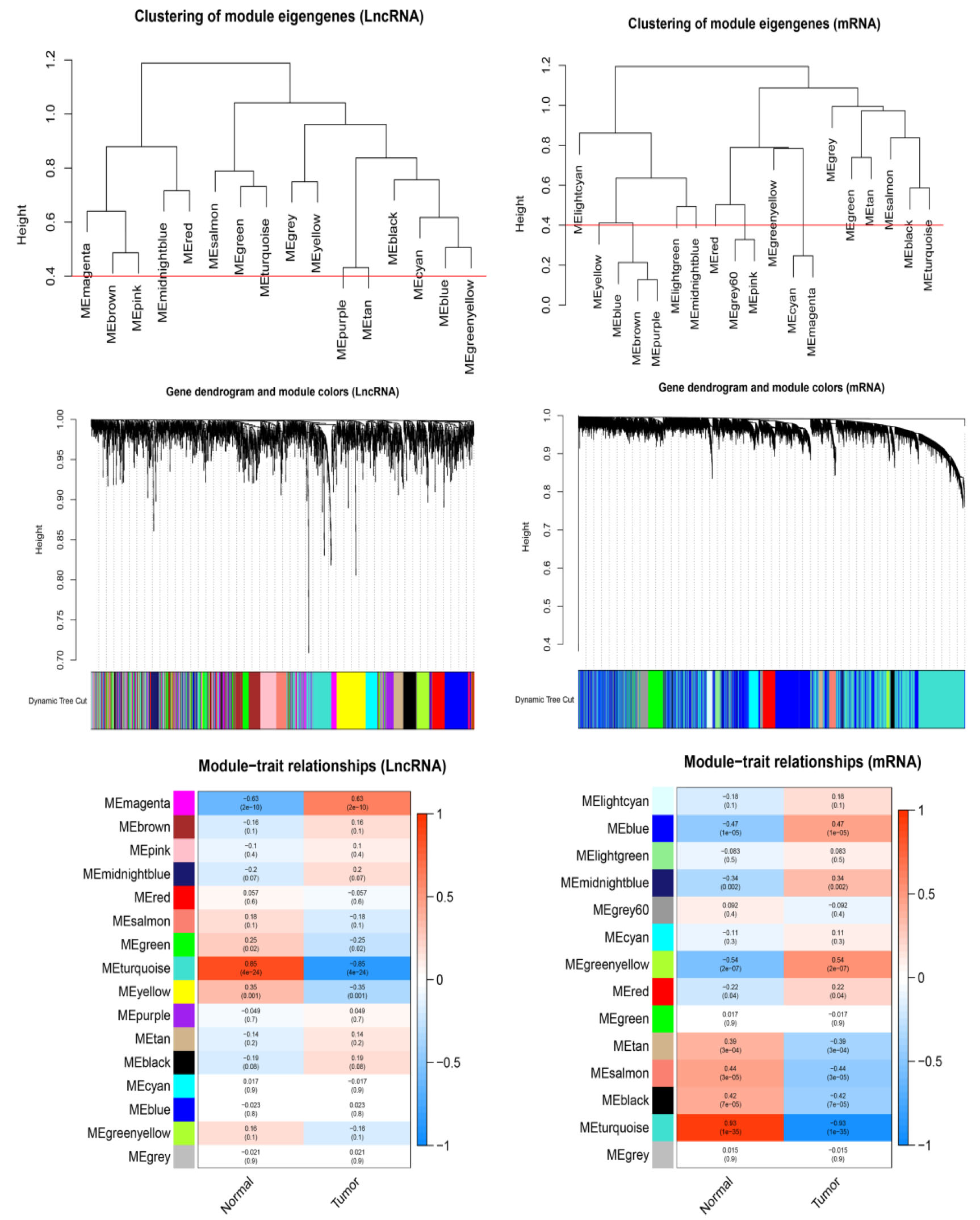
Our research found that under the conditions of |LogFC|
Differential LncRNAs
and mRNA expression in GEO
Differential LncRNAs
Module structure and connections in the expressed data can be visualized by using the WGCNA R software package. As the Fig. 3 shown, we found 16 co-expressed lncRNA modules and 14 co-expressed mRNA modules, after merge similar modules. In module-trait relationships of LncRNA, we found the magenta module displayed highest relationship with tumor (
Differential expression genes pairwise.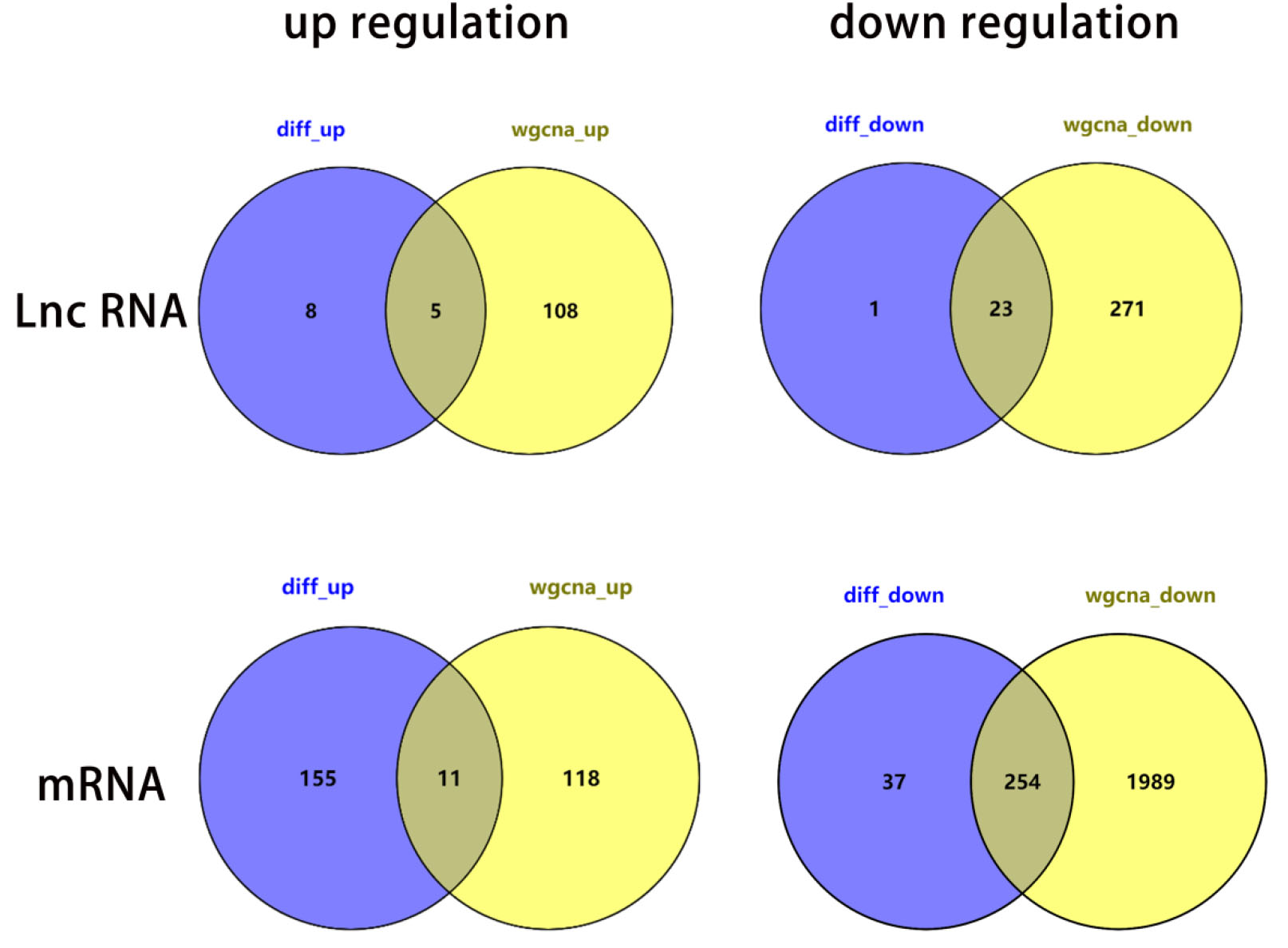
We found there were 6 LncRNAs (CCDC39, FAM182A, SRGAP3-AS2, ADAMTS9-AS2, AC020907.2, SFTA1P) were picked up and set the network (Fig. 5) with 12 miRNAs (hsa-miR-107, hsa-miR-125b-5p, hsa-miR-129-5p, hsa-miR-1297, hsa-miR-135a-5p, hsa-miR-17-5p, hsa-miR-206, hsa-miR-20b-5p, hsa-miR-23b-3p, hsa-miR-27a-3p, hsa-miR-338-3p, hsa-miR-363-3p) which connected to 12 mRNAs (SEMA6D, LOXL2, FGFR2, KLF4, NFIA, LDLR, DPYSL2, SH3BP5, MCC, GATA6, CRIM 1, SPRY2). In the network, it contains 56 connections, green indicates down-regulated LncRNAs, red indicates up-regulated mRNAs, blue indicates down-regulated mRNAs, and orange means miRNAs. We founded all LncRNAs were down-regulated, only one mRNA (LOXL2) was up-regulated and the remaining 11 were down-regulated.
Competing endogenous RNA network. Green indicates down-regulated LncRNAs, red indicates up-regulated mRNAs, blue indicates down-regulated mRNAs, and yellow means miRNAs.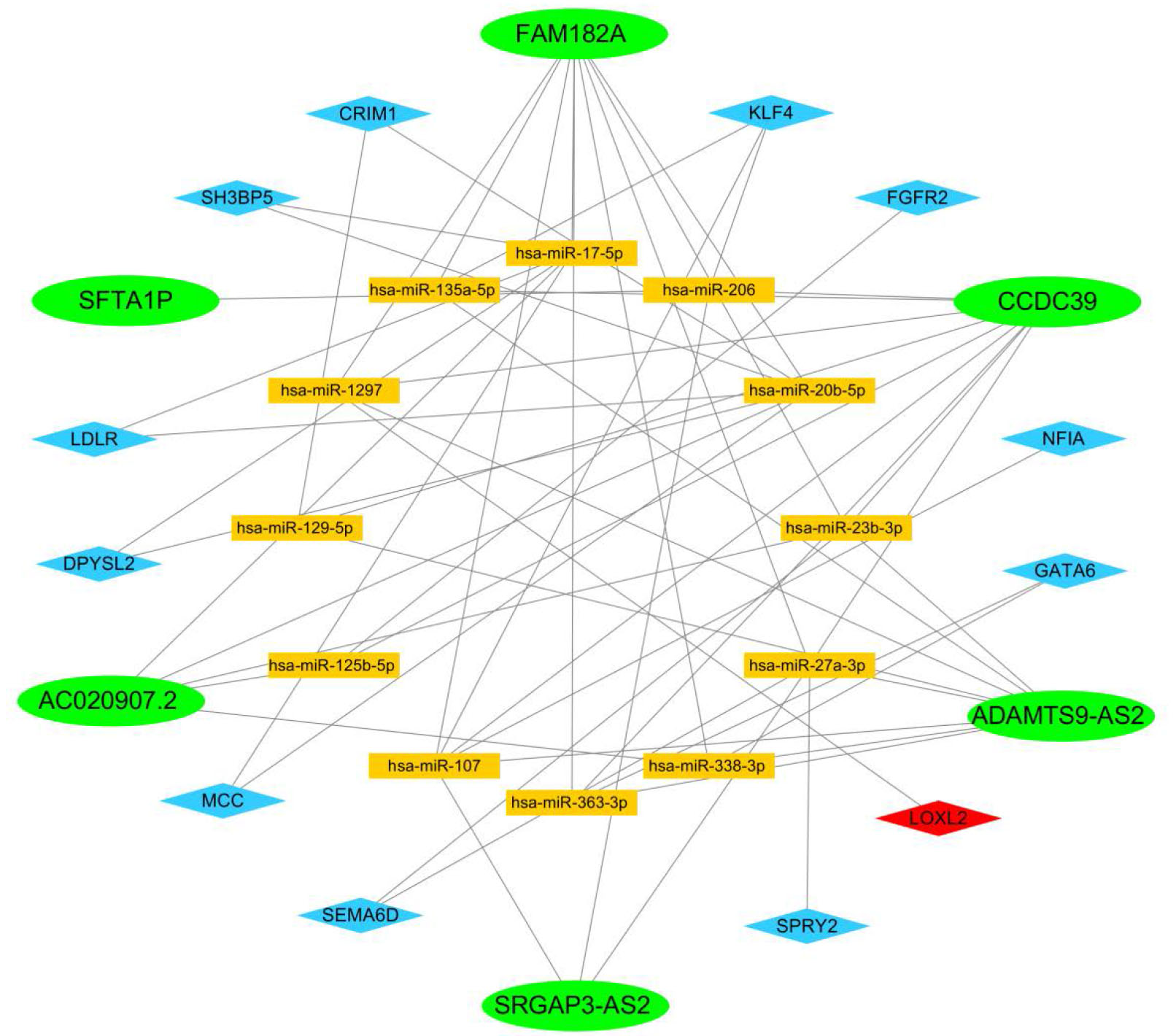
Enrichment analysis. (a) GO enrichment analysis: the first 20 GO terms in the bar graph; 5 GO terms with the smallest adjust 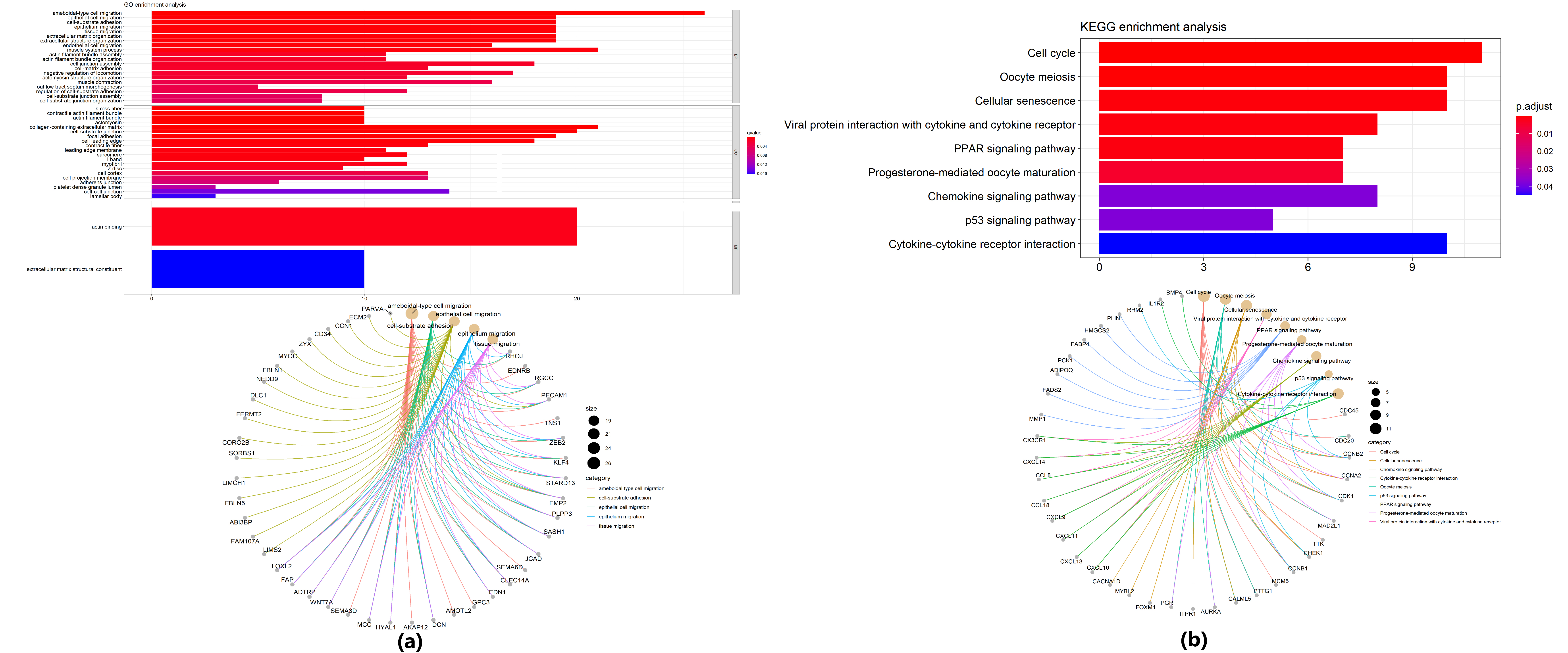
PPI net: line means strength, picture inside shows the structure of protein.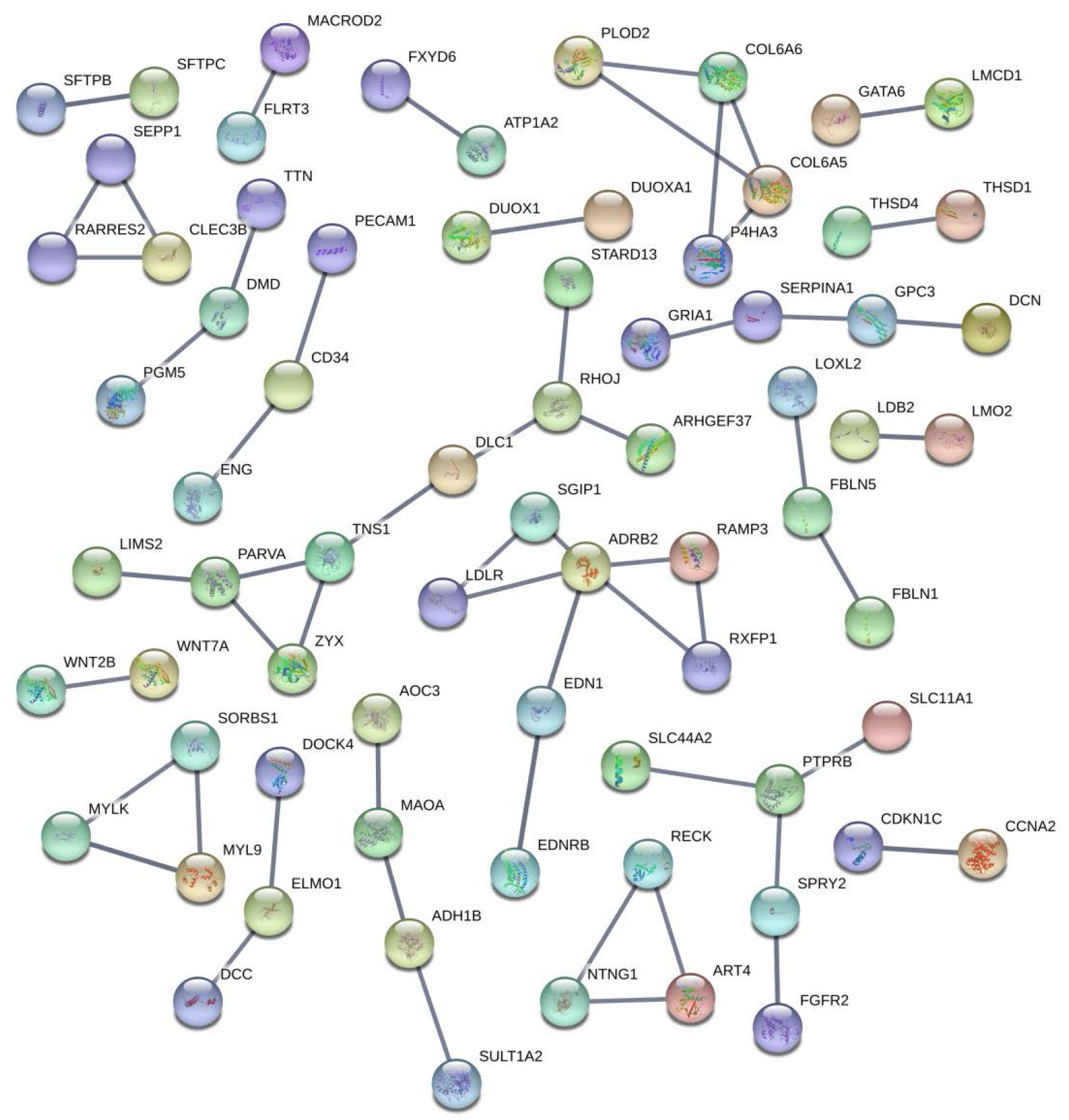
We performed GO enrichment analysis on 265 mRNA in overlapped differential gene expression. In the GO analysis, 103 GO terms of biological process, 22 GO terms of cellular component level localization and 2 GO terms of molecular function were significantly enriched among these mRNAs. According to the adjust
KEGG pathway enrichment analysis
KEGG functional analysis showed that the mRNA in overlapped differential gene expression is involved in the biological process of LAD. The bar chart (Fig. 6) showed after screened from the KEGPATHH_WAY data base, there are 9 signal pathways, including Cell cycle, Oocyte meiosis, Cellular senescence, Viral protein interaction with cytokine and cytokine receptor, PPAR signaling pathway, Progesterone-mediated oocyte maturation, Chemokine signaling pathway, p53 signaling pathway, Cytokine-cytokine receptor interaction. And all of the KEGG functional analysis results were drawn on the circle graph in Fig. 6.
PPI network
There was a total of 71 nodes and 57 edges in the network (Fig. 7). We found EDN1 and EDNRB can interact with each other and this interaction has the highest combined score (0.999). In the PPI network, ADRB2 has the most interaction, including EDN1, LDLR, RAMP3, RXFP1, SGIP1.
Discussion
Lung cancer is a tumor with extremely high incidence and mortality worldwide. Lung cancer treatment methods for different pathological types are also different. With the advent of the era of precision medicine, we have begun to seek personalized therapy at the molecular level. According to a large number of researches on long non-coding RNA in recent years, compared with protein-coding genes and miRNAs, LncRNAs have shown more tissue-, cell type- and cancer-specific expression patterns, and lead to more possibilities in better deciphering molecular heterogeneity of cancer subtypes [26]. Additionally, we performed KEGG pathway enrichment analysis and GO analysis, and respectively, we got 9 biological pathways and 42 genetic functions (including 3 aspects: CC, MF, BP) related to lung cancer that may lead to the therapeutic development of lung adenocarcinoma, which will provide new ideas for targeted treatment of lung cancer in the future. Finally, we constructed the PPI network, which will be important to understand the reaction mechanism of biological signals and energy substance metabolism in lung adenocarcinoma, and the functional connection between proteins as well.
This regulation network we found in our research could provide us with more knowledge of the sophisticated regulation patterns in lung cancer [2].
In our ceRNA network, there were six hub LncRNAs, after we compared them with other researches, we found ADAMTS9-AS2, SFTA1P, and SRGAP3-AS2 have already been found to express differentially and played a vital role in ceRNA network about lung adenocarcinoma in other’s paper before. Hence, our finding is meaningful and has the evidence to support. Besides, these core LncRNAs also have big influence on other types of tumor and diseases [27].
Previous researches about CCDC39 different expression in lung cancer are rare, and the majority of the discussions are about primary ciliary dyskinesia. Primary ciliary dyskinesia (PCD) is a kind of autosomal recessive genetic disease, and most patients normally have the symptoms about the respiratory system, such as neonatal respiratory distress of unknown cause, daily wet cough [28]. According to one of the researches containing a cohort of 59 patients who came from 54 primary ciliary dyskinesia families, they found 12 families had the CCDC39 mutation. And all the changes consisted of frameshift, nonsense and essential splice site mutations [29]. Besides, in some animal experiments, CCDC39 mutation has also proved to cause the neonatal hydrocephalus with abnormal motile cilia development in mice [30] and is important to assembly of inner dynein arms and the dynein regulatory complex in dogs [31].
The differential expression of ADAMTS9-AS2 has been shown to have a deep impact on the discovery and development of lung cancer. A study shows ADAMTS9-AS2 was low expression in lung cancer tissues by qRT-PCR verification, and when it was highly expressed in lung cancer cells, it would reduce the proliferation ability and inhibit migration of them. In addition, by the results of cell cycle assay, they found this gene could restrict the lung cancer cells’ development. Apart from this, they built a Tumor xenograft model to analyze this LncRNA’s effect on the development of lung cancer. After a month later, they measured the weight of tumor and found ADAMTS9-AS2 constrained the growth of lung tumor in vivo [32]. Different expression of ADAMTS9-AS2 has also imposed significant influence on other tumors including ovarian cancer (OA) [33], esophageal cancer [34], colorectal cancer [35], and gastric cancer (GC) [36]. In a study about the function of it in OA, they think overexpression of ADAMTS9-AS2 may contribute to reducing ovarian cancer cells’ proliferation, invasion, epithelial-mesenchymal transition (EMT) processes and also constrained its growth, which linked to mortality [33]. This research may provide us with an idea that the process of lung cancer is similar to the process of OA. Based on these evidences, we could infer that ADAMTS9-AS2 down-regulation is one of the main reasons leading to the tumorigenesis and development in LAD. Thus, we can predict it can be used as a biomarker in lung cancer in the future.
This is not the first time to find the down-regulation of SRGAP3-AS2 in LAD. In a previous investigation, with using quantitative real-time PCR in cells and tissue specimens from patients in experimental hospital, they detected the expression levels of 8 different regulation lncRNAs. Then, after compared with microarray data, they confirmed that SRGAP3-AS2 was down-regulated in LAD [37]. This result is the same as ours, which will support our findings.
SFTA1P is different-expression in both lung adenocarcinoma and lung squamous cell carcinoma (LUSC) by analyzing the genetic date from databases and LUSC samples. But it was different in these two different pathological types, it is up-regulation in LUSC [38] and shows opposing expression trend in LAD [39], with low expression. Therefore, it can be used as a histochemical marker to distinguish these two types. Furthermore, the higher this gene expression, the worse the prognosis of LUSC has [40]. By performing the Kaplan-Meier survival analysis, migration, invasion assays and in vivo studies, they verified the tumor suppressor role of SFTA1P in non-small cell lung cancer: SFTA1P could suppress LUAD cells’ migration and invasion in vitro, inhibit LUAD cell metastasis in vivo and have great impact on LUAD cell proliferation [39]. Besides, in recent studies, SFTA1P differently expressed in many kinds of tumor, such as hepatocellular carcinoma [41], gastric cancer [42] and oral squamous cell carcinoma [43].
Conclusion
In summary, we downloaded four gene chips for lung adenocarcinoma from the GEO database, and after analyzing it, we found statistically significant differential expression of LncRNAs and mRNAs through two different means, and established a ceRNA network and PPI net. By GO and KEGG pathway enrichment analysis, we made the investigation of the molecular mechanisms of lung adenocarcinoma’s pathogenesis and progression. This study provides a new direction for basic and clinical research related to LAD, and is expected to provide new targets for early diagnosis, prognostic evaluation and clinical treatment of lung cancer [44].
Footnotes
Acknowledgments
This work was supported by the grants from China Medical University and National College Student Innovation and Entrepreneurship Training Program.
Conflict of interest
The authors declare that they have no conflict of Interest.
Human participants and animal rights
This article does not contain any studies with human participants or animals performed by any of the authors.