
Abstract
BACKGROUND:
Invasive breast cancer is a highly heterogeneous tumor, although there have been many prediction methods for invasive breast cancer risk prediction, the prediction effect is not satisfactory. There is an urgent need to develop a more accurate method to predict the prognosis of patients with invasive breast cancer.
OBJECTIVE:
To identify potential mRNAs and construct risk prediction models for invasive breast cancer based on bioinformatics
METHODS:
In this study, we investigated the differences in mRNA expression profiles between invasive breast cancer and normal breast samples, and constructed a risk model for the prediction of prognosis of invasive breast cancer with univariate and multivariate Cox analyses.
RESULTS:
We constructed a risk model comprising 8 mRNAs (PAX7, ZIC2, APOA5, TP53AIP1, MYBPH, USP41, DACT2, and POU3F2) for the prediction of invasive breast cancer prognosis. We used the 8-mRNA risk prediction model to divide 1076 samples into high-risk groups and low-risk groups, the Kaplan-Meier curve showed that the high-risk group was closely related to the poor prognosis of overall survival in patients with invasive breast cancer. The receiver operating characteristic curve revealed an area under the curve of 0.773 for the 8 mRNA model at 3-year overall survival, indicating that this model showed good specificity and sensitivity for prediction of prognosis of invasive breast cancer.
CONCLUSIONS:
The study provides an effective bioinformatic analysis for the better understanding of the molecular pathogenesis and prognosis risk assessment of invasive breast cancer.
Keywords
Abbreviations
Background
Breast cancer is the most commonly diagnosed cancer and the leading cause of cancer-related death (11.6% of the total cancer deaths up to 2018) in women [1]. Although many tumor biomarkers and target genes have been associated with breast cancer, the incidence is increasing and prognosis still remains unfavorable, mainly owing to the late diagnosis and limited management options [2]. The available clinical information has limited predictive power because of the complex molecular mechanisms of tumor regulation. Most studies have used a single biomarker for the prediction of prognosis of patients with breast cancer. Therefore, it is necessary to develop a new model based on several biomarkers for the more accurate prediction of the survival of patients with breast cancer.
Next-generation sequencing technology has been widely used in the diagnosis, classification, targeted therapy, and prognosis of cancers [3, 4]. The recent advances in oncology bioinformatics have led to the development of drugs that target the signaling pathways in cancer cells and stroma and vasculature in tumor tissues, greatly extending the survival of patients [5]. In the post-genome era, the main challenge was to use system biology methods to mine and verify new tumor-associated biomarkers from multi-study data, thereby leveraging large amounts of genomic data to improve the clinical treatment of tumors. Long et al, constructed a prognostic model of liver cancer using four genes (CENPA, SPP1, MAGEB6, and HOXD9) through the integrated analysis of RNA sequencing data. Further analysis revealed the independent prognostic ability of this model in association with other clinical features [6]. Shi et al, identified the expression of 31 long non-coding RNAs (lncRNAs) in tumor tissues as a risk indicator for lung cancer treatment and validated the specificity and sensitivity of the model. This exploratory analysis provides new insights into the identification of the potential prognostic factors [7], thereby demonstrating that these novel next-generation sequencing approaches and data may reveal clinical prognostic biomarkers for cancer. Therefore, we used high-throughput sequencing data to construct biomarker models for the prediction of the prognosis of patients with breast cancer.
Here, we explored the differences in mRNA expression profiles between invasive breast cancer and normal breast. As RNA sequencing gradually replaces microarrays as a preferred transcriptomic platform [8], The Cancer Genome Atlas (TCGA) is a cancer database based on RNA sequencing. We analyzed the RNA expression profiles of 1,096 invasive breast cancer tissues and 112 non-invasive breast cancer tissues. Furthermore, the functions of the differentially expressed mRNAs were determined and a new model was constructed to predict the prognostic survival of patients with invasive breast cancer using several candidates.
Methods
Data processing
The mRNA expression profile data and corresponding clinical information from patients with invasive breast cancer were obtained from TCGA database. The mRNA expression profile data were derived from 1,096 invasive breast cancer tissue samples and 112 normal tissue samples. After filtering and excluding incomplete clinical data as well as the data showing no correlation between expression profiles and overall survival (OS), a total of 1,076 invasive breast cancer samples were retained for the construction of the prognostic risk model. Data from TCGA database are publicly available. Therefore, no local ethics committee approval is required.
Screening of differentially expressed mRNAs between invasive breast cancer tissues and non-cancerous tissues
We obtained raw data from TCGA database for mRNA expression profiles associated with invasive breast cancer. The downloaded mRNA data were normalized and differentially analyzed with edgerR package, and the differentially expressed mRNAs were obtained with log 2-fold change and corresponding
Gene ontology (GO) and pathway enrichment analysis of differentially expressed mRNAs
To gain insight into the biological functions of these differential mRNAs, the annotation, visualization, and comprehensive discovery database DAVID v6.8 (
Specific baseline clinical characteristic of 1076 invasive breast cancer patients
Specific baseline clinical characteristic of 1076 invasive breast cancer patients
The volcano diagram about differentially expresses mRNAs between invasive breast cancer tissue and normal tissue samples. Red dots represent up-regulated mRNA and green dots represent down-regulated mRNA.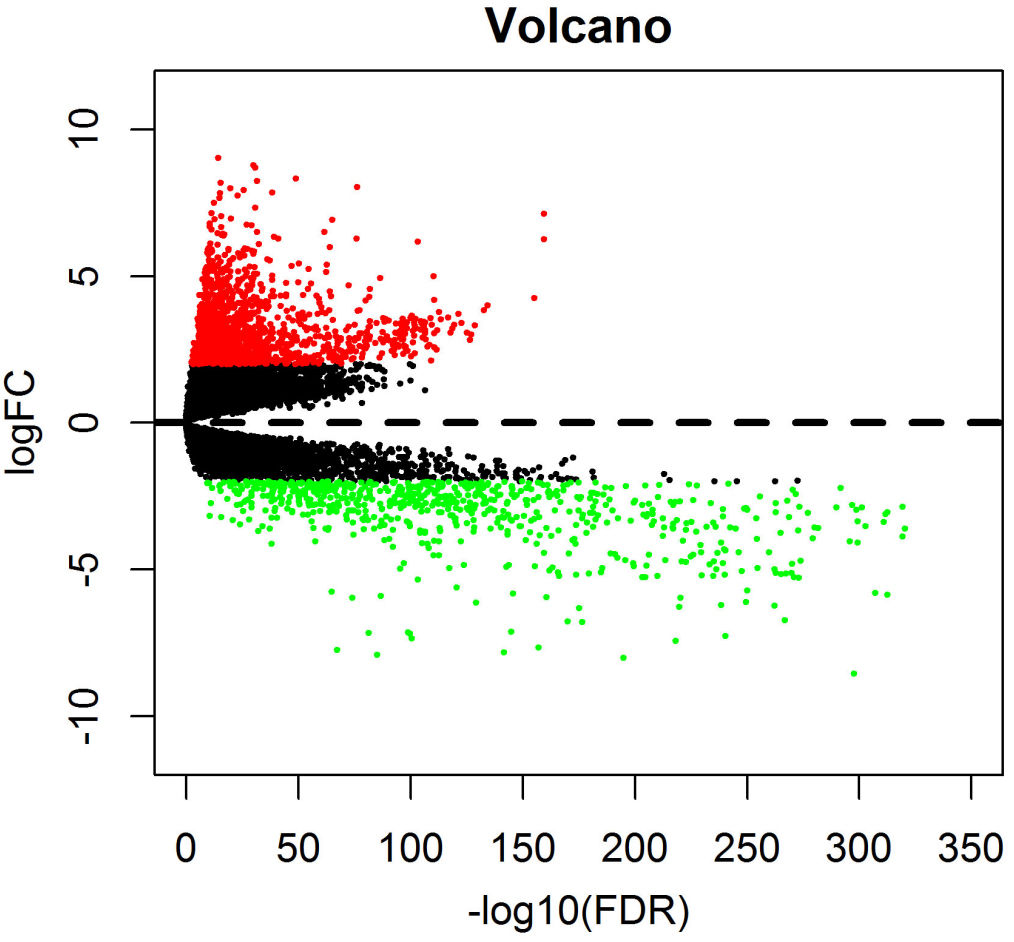
Univariate and multivariate Cox regression analyses were used to study the correlation between patient OS and gene expression level. A value of
Gene ontology analysis of the differentially expressed mRNAs in invasive breast cancer. Top ten terms were selected according to count and
-value
0.05. Count: the number of enriched genes in each term
Gene ontology analysis of the differentially expressed mRNAs in invasive breast cancer. Top ten terms were selected according to count and
Pathways enrichment map of differentially expressed mRNA. KEGG terms were selected according to count 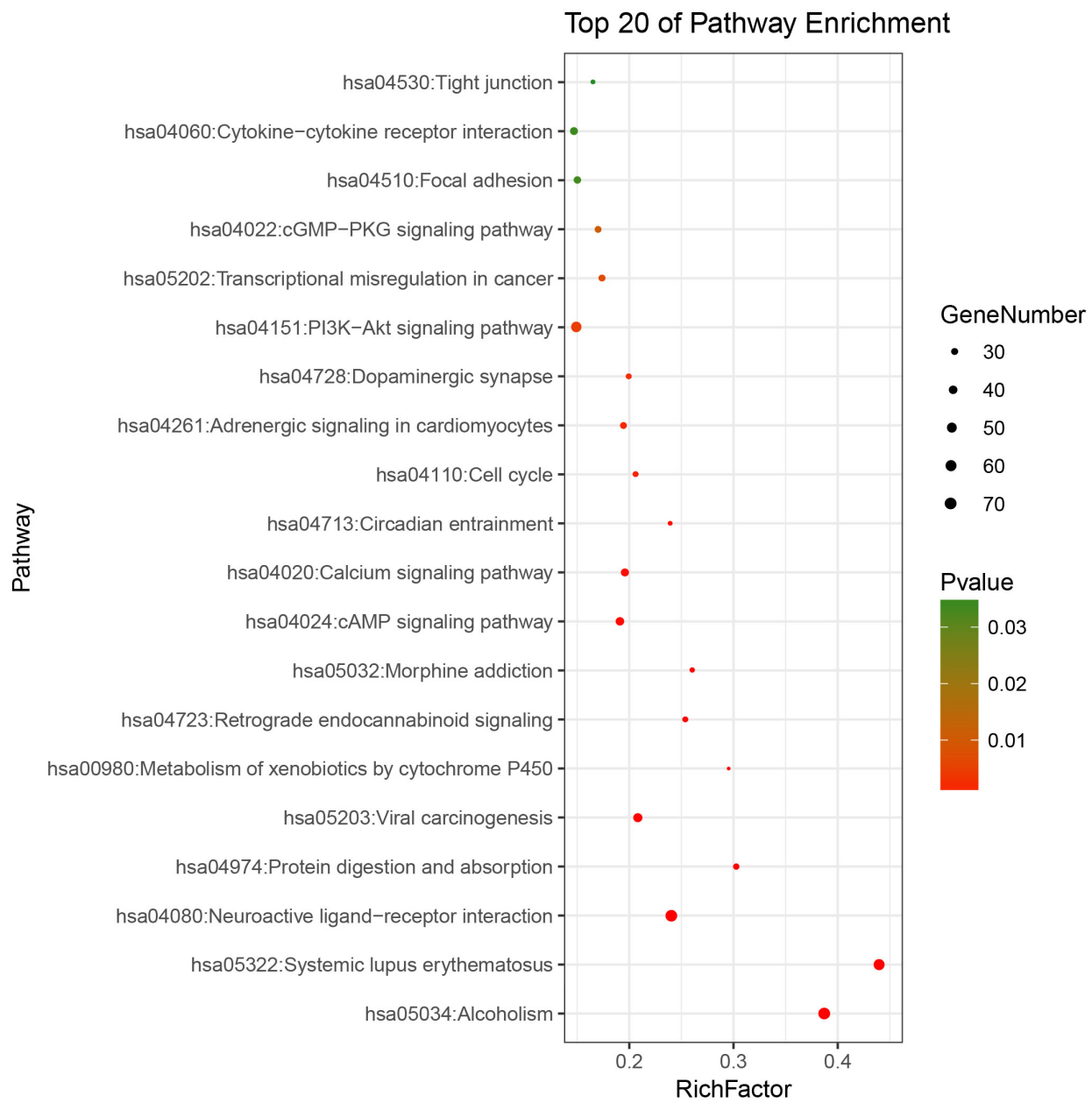
The heatmap of 8 independent invasive breast cancer related prognostic mRNAs. The color from green to red indicates a trend from low to high expression.Taking PAX7 as an example, it can be seen that as the expression of PAX7 increases, the prognostic risk of patients varies from low risk to high risk, indicating that the prognostic risk of patients may be positively correlated with the expression of PAX7.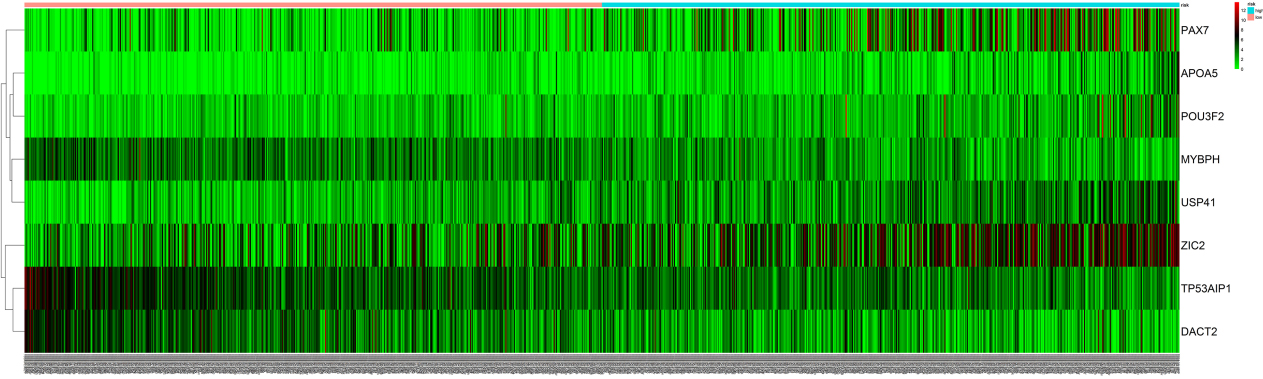
According to the median optimal threshold for prognostic risk, 1,076 patients with breast cancer were divided into low-risk and high-risk groups [11, 12]. In addition, the Kaplan-Meier survival curve method was used to assess OS in patients with high and low risk. Time-dependent receiver operating characteristic (ROC) curve analysis was used to assess the predictive power of the model. We applied an 8-mRNA model to patients with stage I invasive breast cancer to test the validity of the model for survival prediction. In addition, we compared the predictive performance of 8-mRNA model with traditional clinical risk factors (including age, TNM, stage) by univariate and multivariate Cox analysis. First of all, univariate Cox analysis found factors closely related to the prognosis of patients. Then, the effects of many factors on survival time were analyzed at the same time, and the independent prognostic factors could be used to evaluate the survival of patients.
The relationship between 8 mRNAs and the survival of patients with invasive breast cancer.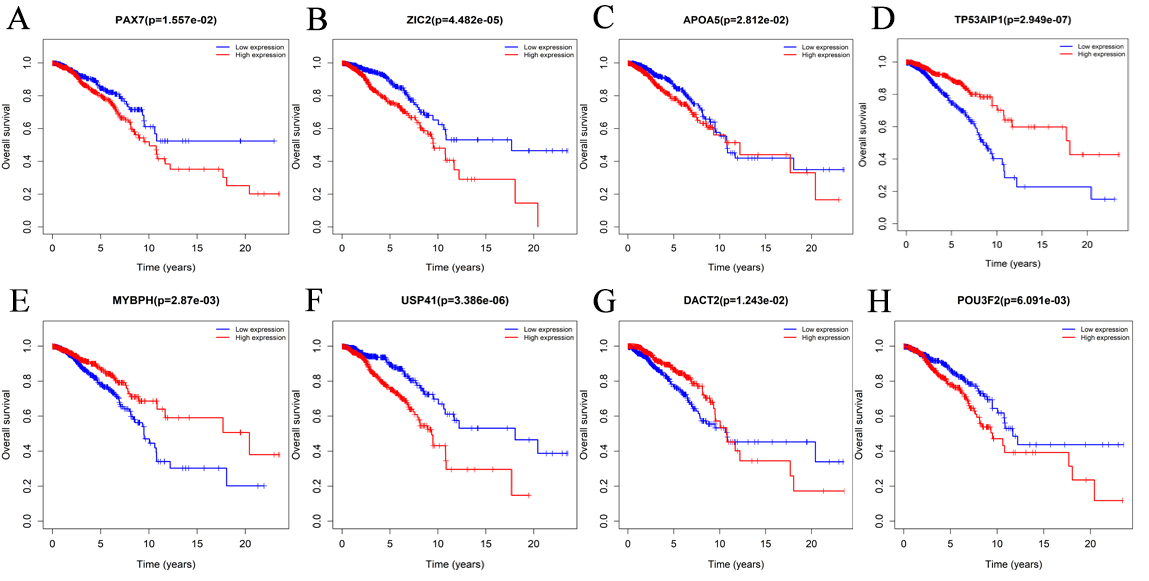
Assessment of prognostic risk in invasive breast cancer patients using an 8-mRNA model based on risk cut-off points.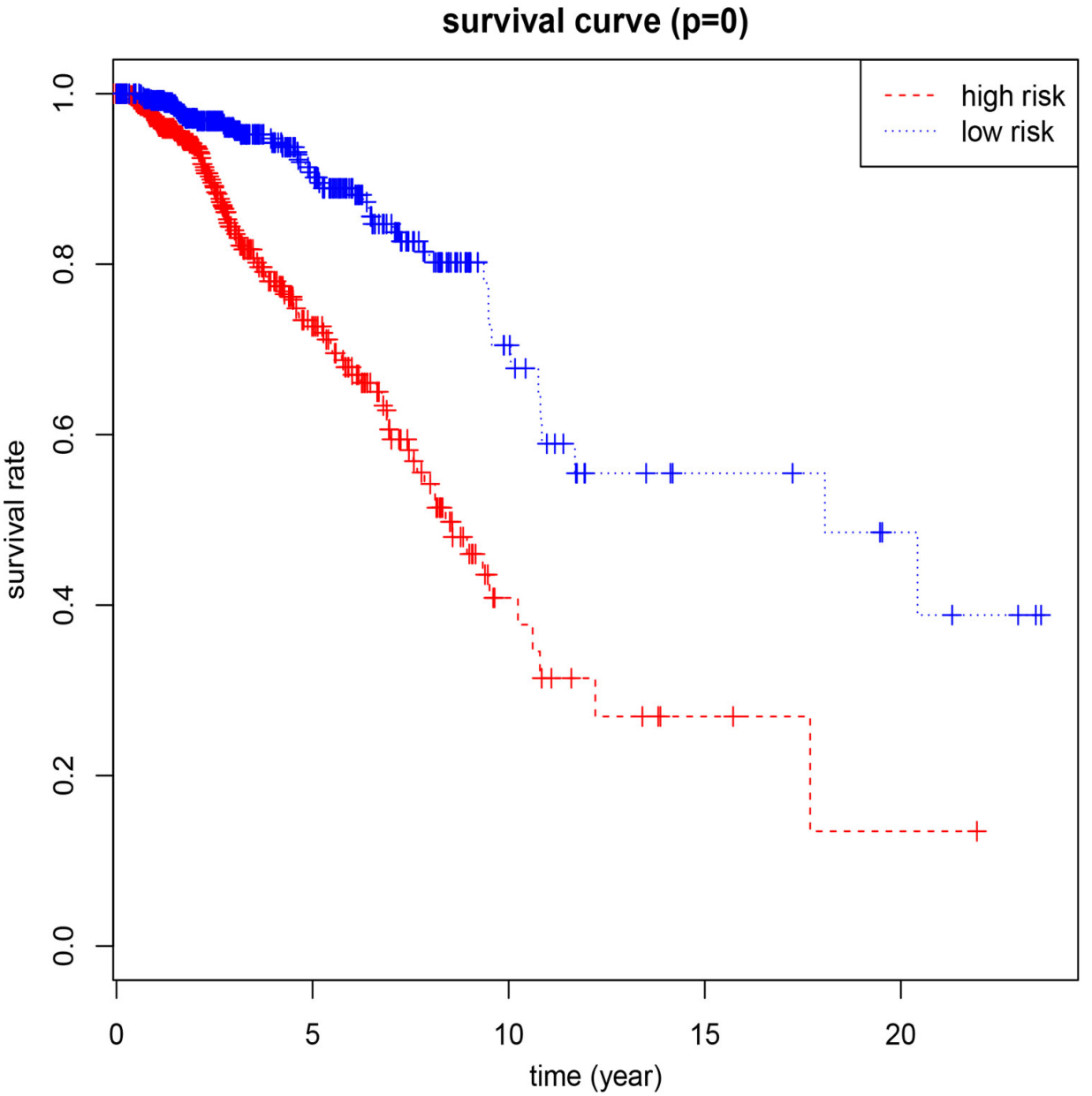
Downloading of TCGA data and differential expression analysis
In this study, 1208 samples were downloaded from the TCGA database and were used to identify differen-tially-expressed mRNAs in breast cancer patients. We analyzed the specific baseline clinical characteristic of 1076 breast cancer patients presented in Table 1 (Supplementary material 1). A total of 2,138 differentially expressed mRNAs (Supplementary material 2), including 1,375 upregulated mRNAs and 763 downregulated mRNAs, were screened using
Functional enrichment and pathway analyses of differentially expressed mRNAs
To understand the functional role of the differentially expressed mRNAs in invasive breast cancer, we performed GO and KEGG pathway enrichment analysis of these mRNAs using DAVID online software. The results indicate that the differentially expressed mRNAs were not only enriched in multiple KEGG pathways but also in molecular functions, biological processes, and cellular components. Pathway analysis revealed that these genes were mainly enriched in the phosphatidylinositol-4, 5-bisphosphate 3-kinase (PI3K)-protein kinase B (Akt) signaling pathway, cytokine-cytokine receptor interaction, calcium signaling pathway, cell cycle, and focal adhesion (Fig. 2). In addition, the results of GO analysis highlighted the enrichment of these genes in biological processes such as transcription from RNA polymerase II promoter, positive regulation of cell proliferation, cell-cell signaling, and cell adhesion; molecular functions such as various binding processes, including sequence-specific DNA binding and calcium ion binding, protein heterodimerization activity, and structural molecule activity; and cellular components such as plasma membrane, integral component of plasma membrane, proteinaceous extracellular matrix, and extracellular matrix (Table 2).
Construction and analysis of the prognosis risk assessment model of differentially expressed mRNAs in invasive breast cancer
We performed univariate Cox regression analysis to study the association between the differentially expressed mRNAs and OS in patients with invasive breast cancer and identified 11 mRNAs that were significantly associated with OS in patients with invasive breast cancer at
11 prognosis-related genes obtained based on univariate Cox regression analysis (
0.001)
11 prognosis-related genes obtained based on univariate Cox regression analysis (
Time-dependent ROC curve analysis of 8-mRNA model for survival prediction of invasive breast cancer patients. (A) at 1 years of OS (AUC 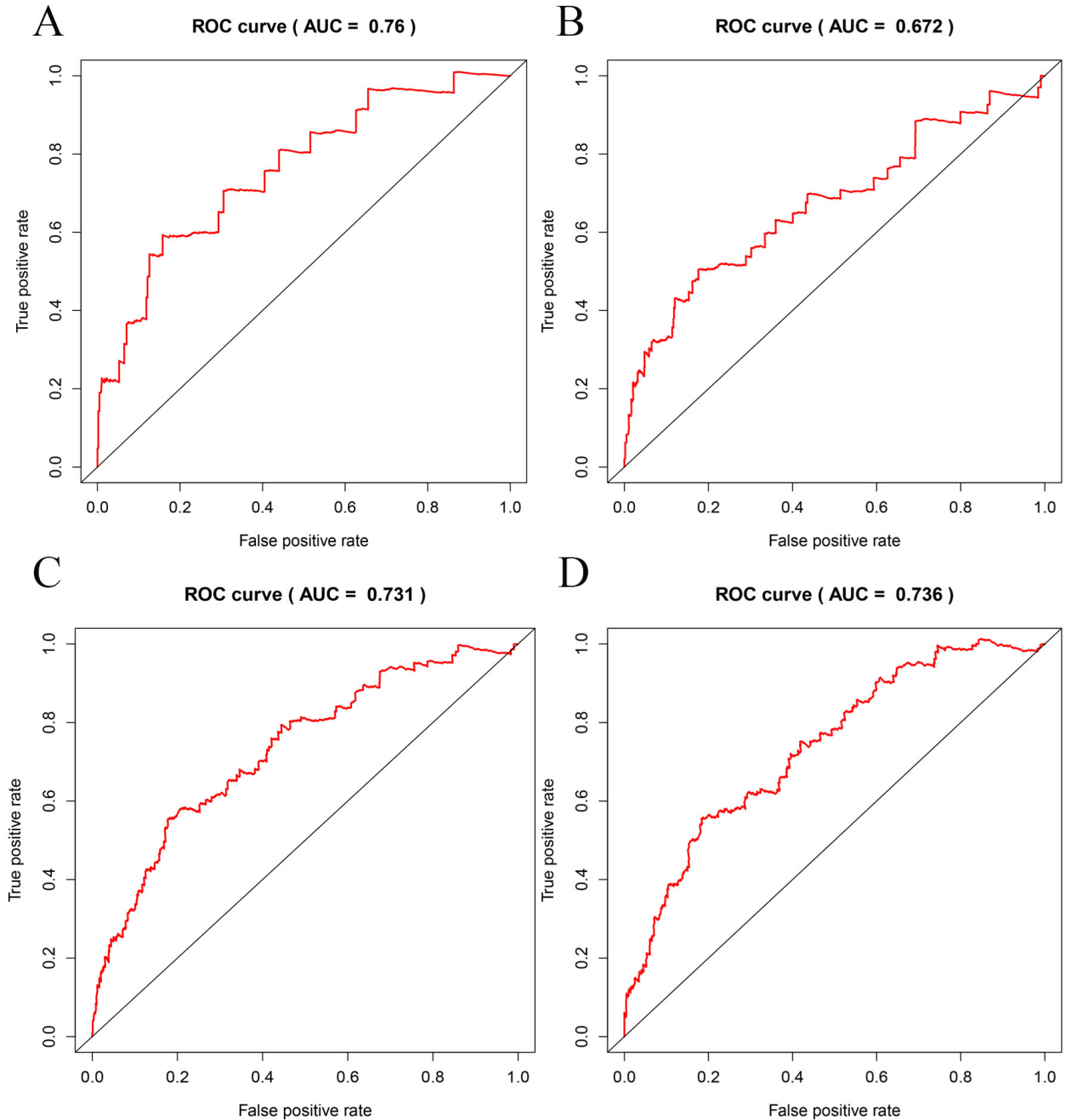
Prognostic index (PI)
With the median PI (value
We compared the predictive performance of the 8-mRNA model with conventional clinical risk factors, including age, TNM, Stage. Univariate analysis found that age, Stage, TNM stage, and predictive performance of the 8-mRNA model were closely related to prognosis (Fig. 7A). Further multivariate analysis found that predictive performance of age and 8-mRNA models could be used as independent prognostic factors to assess patient outcomes (Fig. 7B).
Specific baseline clinical characteristic of 180 patients with stage I invasive breast cancer
Specific baseline clinical characteristic of 180 patients with stage I invasive breast cancer
Univariate (A) and multivariate (B) analysis of clinic pathologic factors for overall survival of invasive breast cancer patients from TCGA.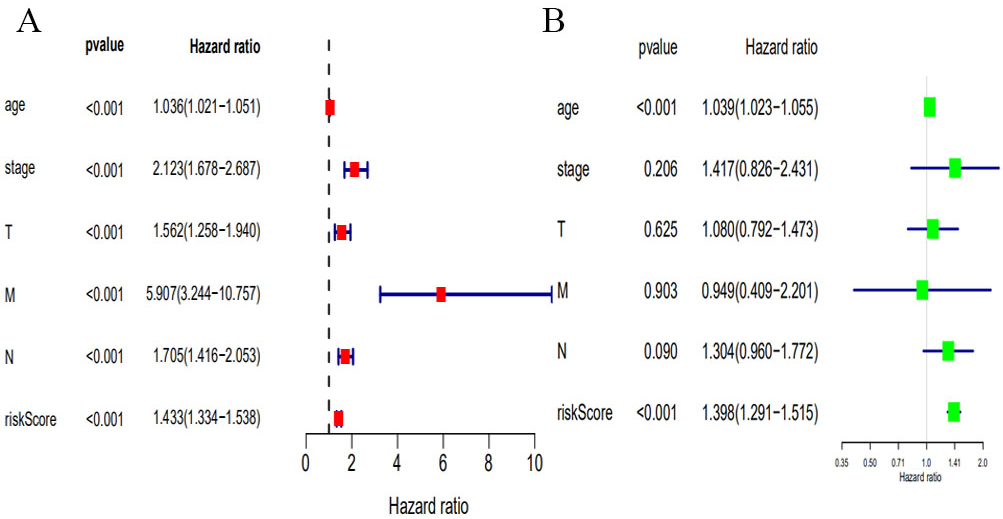
Evaluation of stage I invasive breast cancer patients using 8-mRNA model, (A) assessment of prognostic risk analysis (B) time-dependent ROC curve analysis (the AUC was 0.773 at 3 years of OS).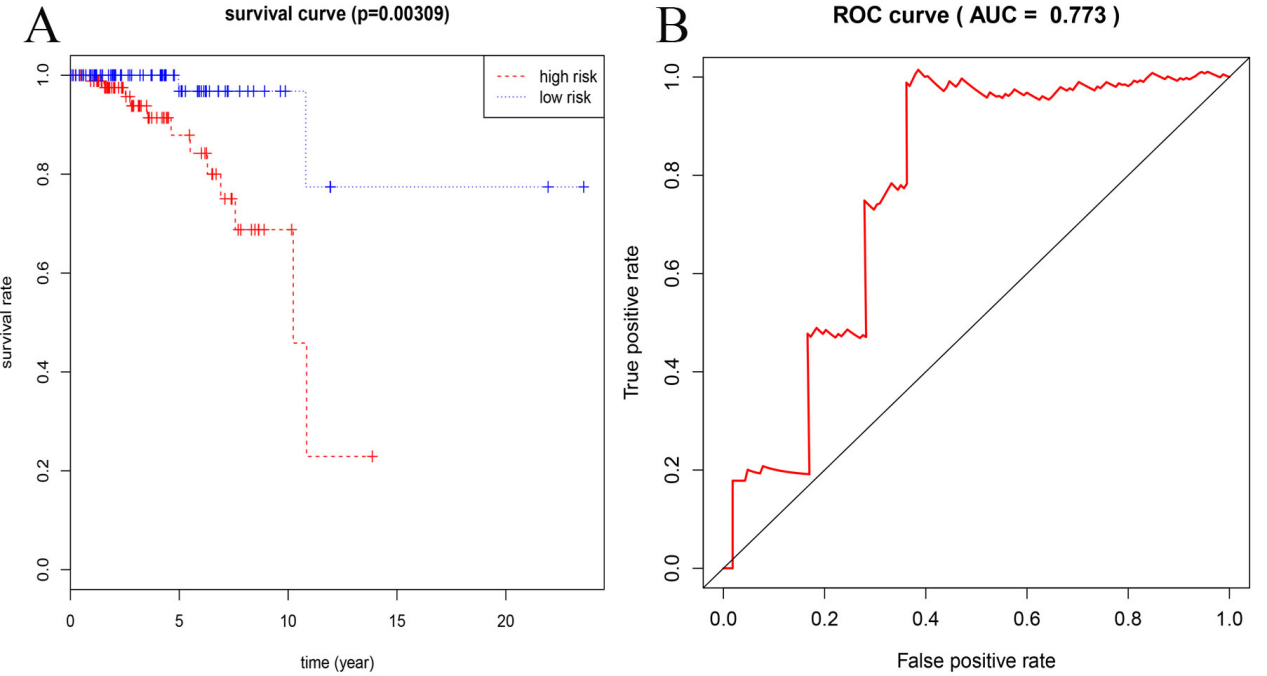
To confirm the validity and sensitivity of the 8-mRNA model for predicting survival, we applied this model to patients with stage I invasive breast cancer for survival risk assessment. We analyzed the specific baseline clinical characteristic of 180 patients with stage I invasive breast cancer presented in Table 4. We used the median risk score (value
Discussion
Breast cancer remains one of the deadliest malignancies in the world, owing to cellular heterogeneity and complex molecular regulatory mechanisms. Therefore, bioinformatic study of breast cancer may provide clinicians with new tools to predict disease prognosis and identify the potential and valuable mRNAs to improve clinical outcomes in patients with breast cancer. In this study based on a large sample of patients with invasive breast cancer from TCGA database, we identified 8 mRNAs, namely PAX7, ZIC2, APOA5, TP53AIP1, MYBPH, USP41, DACT2, and POU3F2. The expression patterns of these 8 mRNAs were significantly associated with OS in patients with invasive breast cancer. The survival of patients was predicted using the 8-mRNA combination model, which is better than the single mRNA and other predictive models. The AUC of the ROC curve predicting the prognosis of patients with invasive breast cancer with 1-, 2-, 3-, and 5-year survival rates were 0.76, 0.672, 0.731, and 0.736, respectively, indicating that the 8-mRNAs prediction model have good effects in survival prediction. The mRNA-based prognostic model for breast cancer may be applied in clinics and the clinicians may classify patients into high-risk and low-risk groups based on the predicted outcomes. For patients in the high-risk group, strategies for frequent monitoring of various tumor indicators should be used, including regular detection of tumor markers and regular chest and abdominal computed tomography examinations for early prevention and diagnosis of breast cancer recurrence, so as to play a role in predicting risk models. In addition, the average value of a single mRNA in the model is correlated with the prognosis of invasive fracture, and can act as a tumor biomarker for invasive breast cancer.
The rapid development of high-throughput sequencing technology and bioinformatic tools has improved our understanding of the molecular regulation mechanisms and characteristics of breast cancer [13]. Alfarsi et al., used the METABRIC dataset to assess kinesin family member 18A (KIF18A) expression at the genomic level and found that the high KIF18A expression has prognostic implications for the prediction of poor endocrine therapy outcomes in patients with estrogen receptor positive breast cancer [14], it shows the potential of mRNA to participate in the clinical treatment of breast cancer. MRNAs may perform the role of a tumor suppressor or an oncogene involved in cancer progression and metastasis and may be used as potential biomarkers for cancer. These molecules offer significant advantages as biomarkers for diagnosis and prognosis [15, 16]. We have developed a prognostic model for breast cancer that includes 8 mRNAs closely related to OS of patients with breast cancer; some of these mRNAs were previously shown to be potential biomarkers.
Paired box gene 7 protein (PAX7) is a DNA-active transcription factor that plays a role in muscle production by regulating the proliferation of muscle precursor cells. In addition to the proven PAX7 expression as a marker of skeletal muscle differentiation in rhabdomyosarcoma [17], PAX7 has been recently used as a highly sensitive marker for Ewing sarcoma [18, 19]. Progress and systemic effects of breast cancer Related, including restricted function and sarcopenia, Wang et al, found that breast cancer progression is associated with the expression of the skeletal muscle stem/satellite-specific transcription factor PAX7. The cytokine-inducible transcription factor NF-
ZIC2, a member of the human zinc finger of the cerebellum (ZIC) family genes, acts as a transcriptional activator or repressor and promotes cell proliferation and migration. ZIC2 dysregulation contributes to the infinite growth of cancer cells, and studies have shown that it can be involved in the pathogenesis of a variety of malignant tumors. The level of ZIC2 complex may be used for the diagnosis and prognosis of patients with hepatocellular carcinoma [21]. ZIC2 plays an indispensable role in the regulation of cell proliferation and apoptosis during the development of pancreatic ductal adenocarcinoma [22]. In addition, ZIC2 acts as a regulatory target for microRNAs. Wang et al., found that miR-129-5p may inhibit cervical cancer by targeting ZIC2 to prevent angiogenesis and suppress cell migration and invasion [23]. Zhang et al., used the luciferase reporter gene assay, reverse-transcription qPCR, and western blotting to show that miR-1284 overexpression inhibits ZIC2 protein expression in breast cancer cells. In addition, ZIC2 knockdown inhibits the proliferation, migration, and invasion of breast cancer cells [24]. Therefore, ZIC2 may be an effective therapeutic target for breast cancer. In addition, ZIC2 may be used as invasive breast cancer prognostic biomarkers of cancer, the high expression of ZIC2 is related to the poor prognosis of breast cancer.
p53-regulated apoptosis-inducing protein 1 (TP53AIP1) gene is a TP53 target and may play an important role in mediating p53/TP53-dependent apoptosis [25]. In cutaneous malignant melanoma, the TP53AIP1 gene plays a key role by inducing apoptosis in response to UV-mediated DNA damage. Truncated TP53AIP1 mutations tend to cause cutaneous malignant melanoma [26]. Existing studies have shown a variety of drugs that inhibit breast cancer cell invasion through the p53 signaling pathway and may enhance the sensitivity of breast cancer cells to drugs [27, 28]. TP53AIP1 may be involved in the p53 signaling pathway and cause cell cycle arrest and apoptosis in breast cancer cells, and the low expression of TP53AIP1 may lead to poor prognosis of invasive breast cancer.
Myosin-binding protein H (MYBPH) binds to myosin and may be involved in the interaction with thick filaments in the A band. Cell migration driven by actomyosin assembly is a critical step in tumor invasion and metastasis. Hosono et al, found that MYBPH is directly transactivated in lung adenocarcinoma by the thyroid transcription factor 1 (TTF-1) and through the direct inhibition of the non-muscle myosin IIA via phosphorylation of the myosin regulatory light chain (RLC) [29]. MYBPH may inhibit Rho-associated protein kinase 1 (ROCK1) expression and negatively regulate actomyosin tissue; this effect may reduce single cell motility and increase collective cell migration, resulting in reduced cancer invasion and metastasis [30]. Therefore, the expression of MYBPH may be related to invasion, migration, and metastasis of breast cancer, inhibition of MYBPH is associated with poor prognosis of invasive breast cancer.
Dishevelled binding antagonist of beta catenin 2 (DACT2), involved in the regulation of intracellular signaling pathways during development, may act as a tumor suppressor in various tumors and is often downregulated by hypermethylation. DACT2 is involved in the molecular regulation of a variety of tumors, such as colorectal cancer [31], head and neck squamous cell carcinoma [32], non-small cell lung cancer (2014), and liver cancer [33]. Li et al., found that DACT2 inhibits breast cancer cell growth by blocking the G1/S phase transition [34]. In addition, Guo et al., found that the hypermethylation of DACT2 gene promoter contributes to gene loss in breast cancer, demonstrative of its tumor suppressor role [35]. Xiang et al., found that the ectopic expression of DACT2 induces apoptosis of breast cells in vitro and further inhibits breast cancer cell proliferation, migration, and epithelial to mesenchymal transition by antagonizing the Wnt/
The transcription factor POU class 3 homeobox 2 (POU3F2) plays a key role in neuronal differentiation (through similarity) and its expression has been detected in both POU3F2 glioblastoma and melanoma. POU3F2 is involved in tumor carcinogenesis and migration and other carcinogenic features [37, 38]. Chen et al, found that POU3F2 expression is positively correlated with tumor-associated NADH oxidase (tNOX) protein expression, and the overexpression of POU3F2 (with the corresponding upregulation of tNOX expression) enhances the proliferation, migration, and invasion of human gastric cancer cells, indicative of the involvement of POU3F2 in tumorigenesis through the transcriptional regulation of tNOX expression [39]. The high expression of POU3F2 may induce the proliferation, migration, and invasion of breast cancer cells.
Tumor staging is an important parameter, but patients from the same tumor stage may have different clinical outcomes, indicating that the prognosis of patients with breast cancer may not be effectively predicted. To validate the predictive performance of the cancer survival model, we tested the model in patients with stage I invasive breast cancer. The predictive model successfully classified patients with stage I invasive breast cancer into high-risk and low-risk groups, and the results were statistically significant. The results suggest that patients with invasive breast cancer may benefit from the predictive model. Therefore, further experimental research to explore the clinical predictions of the predictive model could provide new insights into OS prediction for patients with invasive breast cancer.
Among the prediction models we have studied, the role of four genetic biomarkers in breast cancer has not been studied and may provide some clinical indications and insights for the identification of prognostic factors for breast cancer. In this study, we obtained invasive breast cancer risk prediction models based on clinical real samples and statistical analysis of high-throughput data, and obtained invasive breast cancer biomarkers not found in the current study. The innovative research provides ideas for the next step of laboratory research and clinical research, and can assist the clinical treatment of invasive breast cancer.
The existing clinical prognosis evaluation system can predict the prognosis of patients based on the clinical and pathological characteristics of patients. The 8-mRNA prediction model can predict the prognosis of patients with invasive breast cancer in advance according to the results of model detection. The patient’s genomic risk score has important predictive power, and can be used in combination with clinical information, so that the prognosis of patients at all pathological stages can be better evaluated, so as to obtain more suitable and accurate treatment. Our prediction method reduces sequencing costs, making the application of targeted sequencing based on specific genes more cost-effective and routine. However, the current research still has some limitations. The quality of the samples in TCGA database is very high, and the sample size is small. Therefore, our predictive model still needs to be validated using large-scale clinical data, and multiple regression modeling methods must be used to further improve the prediction accuracy of this model.
Conclusion
In summary, we used TCGA database of invasive breast cancer samples to develop an OS prediction model for patients with invasive breast cancer, thereby providing new ideas for the prognostic prediction of patients with invasive breast cancer. This may allow clinicians to further improve the prognosis of patients with invasive breast cancer through personalized treatment.
Funding
This work is supported by the grants from National Natural Science Foundation of China (81673799) and National Natural Science Foundation of China Youth Fund (81703915), the funders Changgang Sun conceived and designed the study, and Lijuan Liu performed data analysis.
Footnotes
Conflict of interest
The authors declare that they have no competing interests.