
Abstract
Surgery is the most effective treatment for breast cancer patients. However, some patients developed recurrence and distant metastasis after surgery. Adjuvant therapy is considered for high-risk patients depending on several prognostic markers, and lymphovascular invasion has become one of such prognostic markers that help physicians to identify the risk for distant metastasis and recurrence. However, the mechanism of lymphovascular invasion in breast cancer remains unknown. This study aims to unveil the genes and pathways that may involve in lymphovascular invasion in breast cancer. In total, 108 breast cancer samples were collected during surgery and microarray analysis was performed. Significance analysis of the microarrays and limma package for R were used to examine differentially expressed genes between lymphovascular invasion–positive and lymphovascular invasion–negative cases. Network and pathway analyses were mapped using the Ingenuity Pathway Analysis and the Database for Annotation, Visualization and Integrated Discovery. In total, 86 differentially expressed genes, including 37 downregulated genes and 49 upregulated genes were identified in lymphovascular invasion–positive patients. Among these genes, TNFSF11, IL6ST, and EPAS1 play important roles in cytokine–receptor interaction, which is the most enriched pathway related to lymphovascular invasion. Moreover, the results also suggested that an imbalance between extracellular matrix components and tumor micro-environment could induce lymphovascular invasion. Our study evaluated the underlying mechanisms of lymphovascular invasion, which may further help to assess the risk of breast cancer progression and identify potential targets of adjuvant treatment.
Keywords
Introduction
Breast cancer is the second most common cancer with estimated 1.67 million incident cases during the year 2012 worldwide. Surgery is an effective treatment for early breast cancers. 1 However, some breast cancer patients developed recurrence or distant metastasis after surgery.2,3 Adjuvant therapy for breast cancer is determined according to prognostic factors and the risk of recurrence or distant metastasis. Well-established prognostic factors include age, histological grade, tumor size, axillary nodal status, and the presence of hormone receptor.4–6 Previous studies indicated that patients with small tumors had favorable overall survival than patients with larger tumors. 7 Furthermore, tumor size was correlated with the time to develop metastasis. 8 The presence of estrogen receptor (ER) and/or progesterone receptor (PR) is considered for receiving adjuvant hormone manipulation therapy such as tamoxifen to improve survival.9,10 Human epidermal growth factor receptor 2 (HER2) is also considered a prognostic as well as a predictive marker for trastuzumab responsiveness. 4
Regional lymph node involvement means that a cancer already has the ability to develop distant metastasis, and it is one of the indications for adjuvant treatment.11,12 Lymphovascular invasion (LVI) is an important and early step of tumor metastasis and is defined as the presence of carcinoma cells in lymphatic and blood vessels. A previous study showed that patients with LVI had higher mortality rates than patients without LVI. 13 Several studies indicated that LVI is associated with the risk of axillary lymph node involvement, distant metastases, and recurrence.11,12,14–17 However, the underlying mechanism of LVI in breast cancer is unknown. In this study, we attempted to identify crucial molecules and pathways associated with the presence of LVI in breast cancer.
Materials and methods
Breast cancer samples and microarray analysis
Breast cancer tissues were collected and processed as previously described Huang,18 and was reviewed by and approved by institutional review board of Cathay General Hospital. We obtained informed consent from all patients after explanation of the study. Breast cancer samples from pretreated invasive breast cancer patients (<70 years old) were collected during surgery and processed with RNAlater reagent (Qiagen, Germantown, MD) and stored at −80°C liquid nitrogen. Samples with <10% non-malignant constituents were selected for microarray experiment. Immunohistochemical (IHC) test was performed to determine ER positivity status. HER-2 overexpression (IHC 2+/3+) were determined according to ASCO and CAP guidelines by fluorescence in situ hybridization. Grading was performed based on modified Bloom-Richardson (Nottingham) system. In addition, LVI information was obtained under microscopic inspections. After performing microarray experiment using Affymetrix GeneChip® Human Genome U133 plus 2.0 (Affymetrix, Santa Clara, CA), the data was normalized using robust multichip average (RMA) method Irizarry. 19
Data processing and gene expression calculations
All breast cancer patients were divided into two groups according to clinical LVI-positive (LVI+) and LVI-negative (LVI−) phenotypes. Differentially expressed genes (DEGs) were identified using the significance analysis of the microarrays (SAM) method 20 and the limma package in R (version. 2.15.1). 21 For SAM, statistical comparisons were conducted using a two-sided Student’s t test (p < 0.05) and comparisons of multiples of change (>1.5-fold). The percentage of these significant genes identified by chance was referred to as the false discovery rate (FDR). The FDR was set to 0.05, and a q value was provided as the score of the FDR for each significant gene. For the limma analysis, after normalization and quality control of a gene set, we performed a DEG analysis and selected genes with p < 0.05. Then, common genes between these two statistical methods were selected for further study.
Network construction and pathway analysis
The Ingenuity Pathway Analysis (IPA; Ingenuity® Systems, Mountain View, CA; available at https://www.ingenuity.com) is a web-based software application for integrating, analyzing, and understanding data derived from gene expression measurement. The gene network analysis is used to cluster genes based on their molecular functions and revealed their correlations. We constructed gene networks and relevant metabolic pathways underlying LVI by mapping our genes of interest to the database and subjecting them to a core analysis. In addition, we also consulted Database for Annotation, Visualization and Integrated Discovery (DAVID) bioinformatics tool to find common pathways with the IPA and map the corresponding genes accordingly.
Gene set enrichment analysis and gene ontology
To confirm our finding, gene set enrichment analysis (GSEA) was used to analyze three databases including Kyoto Encyclopedia of Genes and Genomes (KEGG) database, Gene Ontology Biological Processes (GO-BP) database, and Reactome database. This method focuses on groups of genes that share common biological function, chromosomal location, or regulation. 22 We used R clusterProfiler package 23 for analysis. In addition, GO terms were performed to classify their biological processes, molecular functions, or cellular components. We used the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) 24 database to access the GO. We mapped all DEGs and selected GO terms which met statistical significance at p < 0.05 level.
Results
Clinical characteristics of the 108 breast cancer patients
Clinical data of all 108 breast cancer patients are shown in Table 1. Among these cases, most patients showed ER positivity (n = 70, 64.8%) and most of them were diagnosed with pathological stage II (n = 46, 42.6%). At diagnosis, 79 patients (73.1%) were presented with LVI, whereas 29 patients (26.9%) were diagnosed without the presence of LVI and 1 patient (0.9%) had no record for LVI status. Gene expression data were deposited in National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO; submission to GEO in progress).
Clinical characteristics of 108 breast cancer patients.
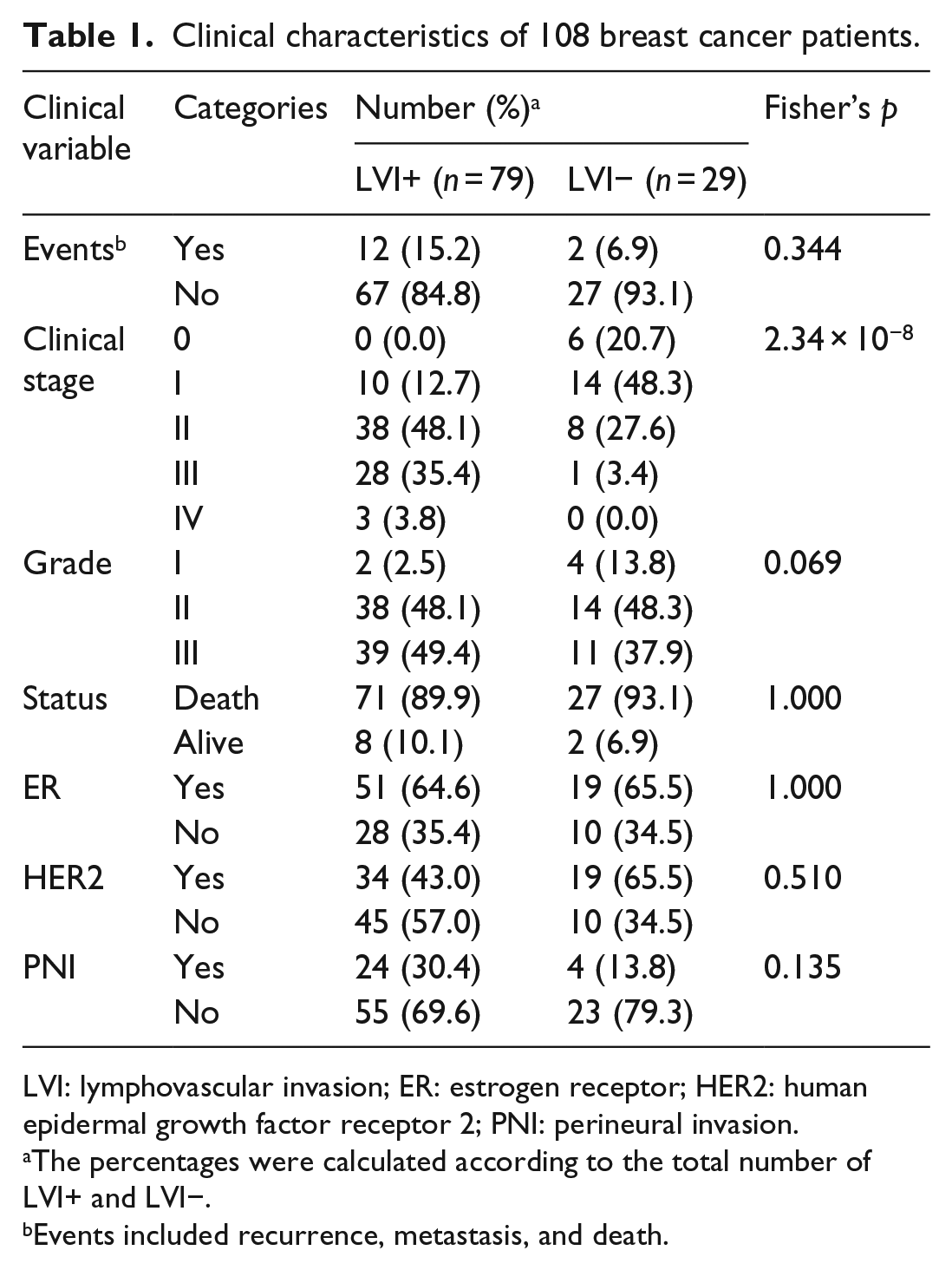
LVI: lymphovascular invasion; ER: estrogen receptor; HER2: human epidermal growth factor receptor 2; PNI: perineural invasion.
The percentages were calculated according to the total number of LVI+ and LVI−.
Events included recurrence, metastasis, and death.
Genes involved in LVI
We used SAM and the limma package to identify DEGs between LVI+ and LVI− samples. The intersection of both statistical methods identified 86 common genes: 37 were downregulated and 49 were upregulated (Supplement Figure 1 and Supplement Table 1).
Protein–protein interaction networks
IPA was used to construct the interaction network of DEGs. We selected human species, breast cancer cell lines, and direct interactions to identify interaction networks. After a core analysis, we found that six networks are involved with filtered DEGs (Supplement Table 3). The connective tissue disorder network was the most significant network related to LVI (Figure 1). In addition, we produced an overlapping network and found that the ubiquitin C (UBC) gene had the highest degree of connectivity to other genes in the network including gene developmental disorders (Figure 2) and nucleic acid metabolism networks (Figure 3).

Network of connective tissue disorder–related genes (red: upregulated; green: downregulated).

Network of developmental disorder–related genes.

Network of nucleic acid metabolism network–related genes.
Pathways associated with LVI
After the IPA core analysis, we found that five significant pathways are related to LVI (Figure 4). The oncostatin M (OSM) signaling pathway was the most enriched pathway associated with LVI. This pathway functions as a part of the cytokine–receptor complex. Interleukin-6 signal transducer (IL6ST) and endothelial PAS domain protein 1 (EPAS1) are two genes that belong to this pathway (Supplement Table 4). Furthermore, we also mapped genes using the KEGG 18 and found similar results of cytokine–receptor interactions being the most enriched pathway associated with LVI (Table 2 and Figure 5). To support our discovery, we further analyzed using GSEA and found that cytokine system was indicated to be the significant biological process associated with LVI in three different databases (Figure 6). In addition to the effect of cytokine–receptor interaction on LVI, we noted that collagen- and lipid-related pathways are also relevant to LVI (Table 2 and Supplement Table 3).

Lymphovascular invasion–related pathways.
Lymphovascular invasion–related pathways using the KEGG analysis.

KEGG: Kyoto Encyclopedia of Genes and Genomes; IL6ST: interleukin-6 signal transducer; TNFSF11: tumor necrosis factor (ligand) superfamily, member 11; ECM: extracellular matrix; COL4A1: collagen alpha-1(IV) chain; COL11A1: collagen alpha-1(XI) chain; LAMA3: laminin subunit alpha 3; PPAPDC1B: phosphatidic acid phosphatase type 2 domain containing 1B; PLA2G7: phospholipase A2 group VII; SNRPE: small nuclear ribonucleoprotein E; TRA2A: transformer 2 alpha homolog; RBM: RNA-binding motif.
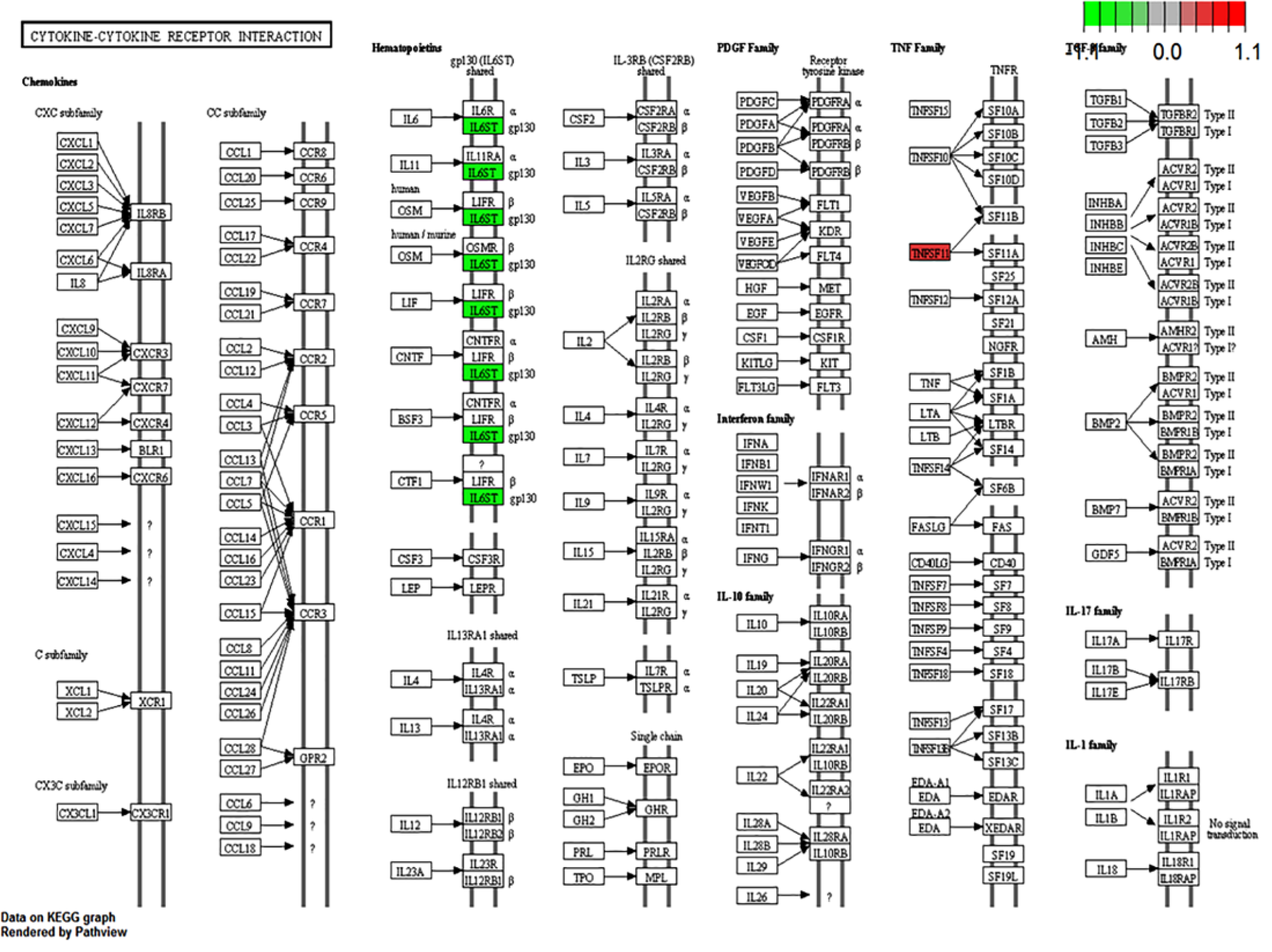
Cytokine–cytokine receptor interaction pathway using the KEGG analysis.

GSEA-identified cytokine system was the significance-enriched pathway associated with LVI.
Subgroup analysis of TNFSF11, IL6ST, and EPAS1
We further assessed the difference in expression levels of tumor necrosis factor (ligand) superfamily, member 11 (TNFSF11), IL6ST, and EPAS1 between LVI+ and LVI− samples in different subgroups. Based on ER status, HER2 status, lymph node metastasis and perineural invasion, permutation analysis was conducted (Supplement Table 2). We found that TNFSF11 and EPAS1 showed a significant correlation with LVI in ER-negative and HER2-positive breast cancer patients. IL6ST and EPAS1 showed a significant correlation with LVI in samples without lymph node metastasis and perineural invasion. These results thus suggest the potential roles of TNFSF11, IL6ST, and EPAS1 as subtype-specific LVI markers.
GO analysis
GO analysis provides information about the functions of the DEGs by classifying them by their biological processes, molecular functions, and cellular components of DEGs. With STRING analysis, we found that biological processes of hemidesmosome assembly, molecular function of receptor binding, and the cellular component of the extracellular region were the most significant biological processes associated with LVI. In addition, development of blood vessels and structural constituents of the cytoskeleton may underpin the phenomenon of LVI (Table 3).
GO enrichment in LVI-positive patients.

GO: gene ontology; LVI: lymphovascular invasion.
Discussion
The aim of this study was to identify genes and pathways associated with LVI in breast cancer. Our study performed a gene expression analysis of Taiwanese breast cancers database and found that the OSM signaling pathway was the most enriched pathway related to LVI in breast cancer. OSM belongs to the interleukin 6 (IL-6) cytokine family and regulates the acute response to infection and injury. 25 Previous studies indicated that OSM plays many crucial roles in cell growth,26–29 bone metabolism,30,31 cholesterol metabolism, 32 and angiogenesis. 33
OSM has been evaluated in several human malignancies. Lilja et al. 34 found that OSM had higher expression in brain tumors than in normal brain tissues. Another study by Fossey et al. 35 showed that OSM induced signal transducer and activator of transcription 3 (STAT3) activation resulting in stimulation of matrix metalloproteinase 2 (MMP-2) and vascular endothelial growth factor (VEGF), finally enhancing invasive behavior and tumor angiogenesis in osteosarcoma. In addition, OSM stimulated mammary tumor metastasis to bone and activated osteoclast differentiation. 36
From IPA analysis, two genes were related to this pathway: IL6ST and EPAS1 or hypoxia-inducible factor 1 (HIF-1). The IL6ST (gp130) functions as a part of the cytokine–receptor complex associated with the IL-6 receptor (gp80). 37 A previous study showed that IL6ST is expressed by human endothelial progenitor cells (EPCs) and is activated by the IL-6 cytokine. The results suggested that IL-6 induced EPC migration, proliferation, and matrigel tube formation via gp80/gp130 activation and STAT3 phosphorylation signaling pathway. 38 Moreover, Yao et al. 39 showed that IL-6 stimulated VEGF-induced angiogenic activity and extracellular signal–regulated protein kinases (ERK1/2) phosphorylation in human cerebral endothelial cells.
EPAS1 showed some identical sequences to HIF-1α, which respond to hypoxia and regulate vascularization. 40 Ke and Costa 41 found that EPAS1 translocates to nuclei and activates genes involved in angiogenesis. Bárdos and Ashcroft 42 also indicated that EPAS1 stimulates the production of VEGF, which is a regulator of tumor angiogenesis, invasion, and metastasis. Moreover, KEGG analysis from our study showed similar results of cytokine–receptor interactions including IL6ST and TNFSF11 being the most enriched pathways associated with LVI.
TNFSF11 is also known as receptor activator of nuclear factor kappa-B ligand (RANKL). RANKL enhanced mammary cell invasion and migration and bone metastasis. 43 In addition, Sfiridaki et al. 44 demonstrated that serum RANKL level was elevated in multiple myeloma and was positively correlated with the potent angiogenic cytokines of hepatocyte growth factor (HGF), VEGF, and IL-6.
Angiogenesis is strongly correlated with tumor progression, invasion, and metastasis.45–47 We surmised that expression alterations of genes in the cytokine–receptor pathway could result in angiogenesis and eventually presented as LVI pathologically.
In addition to cytokine–receptor interactions, we also found that extracellular matrix (ECM) components such as collagens were associated with LVI. Collagens are ECM proteins that play important roles in cell adhesion 48 and stabilization of hemidesmosome assembly. 49 Our study suggested that collagen alpha-1(IV) chain (COL4A1) and collagen alpha-1(XI) chain (COL11A1) were associated with the ECM–receptor pathway. In contrast, previous studies showed that some ECM proteases such as MMP-2 were involved in the progression and lymph node metastasis of gastric cancer, 50 and MMP-9 was involved in the process of neovascularization and angiogenesis.51,52 Our results implied that an imbalance between ECM components and the tumor micro-environment could induce LVI.
From interaction analysis, three networks including connective tissue disorders, developmental disorders, and nucleic acid metabolism were revealed given the filtered genes. UBC was a major protein common to all three networks and had the highest connectivity to others. Although in our dataset, UBC gene did not show the differential expression. We assume that the protein level changes may cause these interactions. Ubiquitin is a small protein covalently attached to the target 53 and generates many thousands of ubiquitinated products. 54 Ubiquitin also plays multiple important roles in cellular processes relevant to cancer.55,56 Ubiquitination can result in several cellular processes including protein degradation 57 and control protein–protein interaction (PPI). 58 Our results suggested that the ubiquitin system interacted with all the networks and may contribute to LVI.
In conclusion, our results with comprehensive bioinformatics approaches including GSEA, GO, PPI, and pathway analyses indicated that the underlying processes of LVI resulted from dysregulations of these three networks. Cytokine–receptor and ECM–receptor interactions are the two major pathways associated with LVI. This study also identified some candidate genes related with LVI which can be potential targets for breast cancer treatment.
Footnotes
Acknowledgements
S.K. and H.S.-C.W. contributed equally to this study.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work was supported by grants from Cathay Medical Research Institute CGH-MR-A-10312; CGH-MR-10208, from the Excellence for Cancer Research Center Grant, funded by the Ministry of Health and Welfare (MOHW105-TDU-B-212-134007) and Cathay General Hospital, Taipei Medical University (No. 105CGH-TMU-05).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.