
Abstract
BACKGROUND:
Metastasis regularly is a marker of the disease development of cancers. Some metastatic sites significantly showed more serious clinical outcomes in non-small cell lung cancer (NSCLC). Whether they are caused by tissue-specific (TS) or non-tissue-specific (NTS) mechanisms is still unclear.
OBJECTIVE:
Explore co-expression gene modules of non-small cell lung cancer metastases.
METHODS:
Weighted Correlation Network Analysis (WGCNA) was used to identify the gene modules among the metastases of NSCLC. The clinical significance of those gene modules was evaluated with the Cox hazard proportional model with another independent dataset. Functions of each gene module were analyzed with gene ontology. Typical genes were further studied.
RESULTS:
There were two TS gene modules and two NTS gene modules identified. One TS gene module (green module) and one NTS gene module (purple module) significantly correlated with survival. This NTS gene module (purple module) was significantly enriched in the epithelial-to-mesenchymal transition (EMT) process. Higher expression of the typical genes (CA14, SOX10, TWIST1, and ALX1) from EMT process was significantly associated with a worse survival.
CONCLUSION:
The lethality of NSCLC metastases was caused by TS gene modules and NTS gene modules, among which the EMT-related gene module was critical for a worse clinical outcome.
Keywords
Abbreviations
The workflow of this study.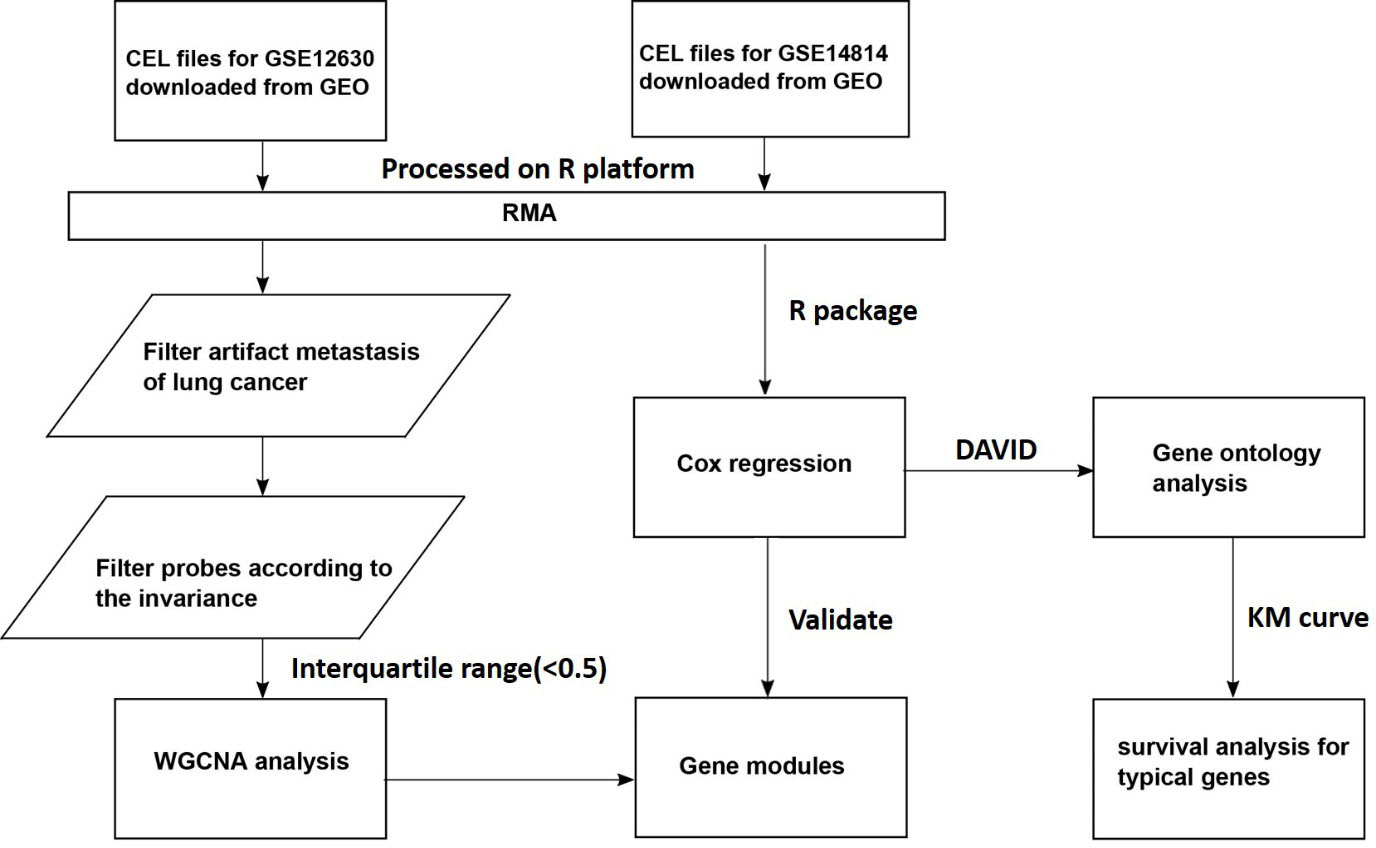
Lung cancer is the most deadly cause of cancer-related deaths worldwide. Among them, non-small cell lung cancer (NSCLCs) accounts for 80%. Along with disease development, metastasis of lung cancer is one of the most significant markers [1, 2]. It is reasonable to hypothesize that there could be some shared mechanism which is responsible for the lethality from the metastases of NSCLC. It has been reported that multiple mechanisms are involved in the process of angiogenesis, hypoxia, circulation, and establishment of a metastatic focus [3].
WGCNA analysis of the metastasis tissues. The upper panel is the hierarchical clustering tree. The lower panel are two heatmaps of the gene module membership from “dynamic tree cut” and “merged” analysis in WGCNA.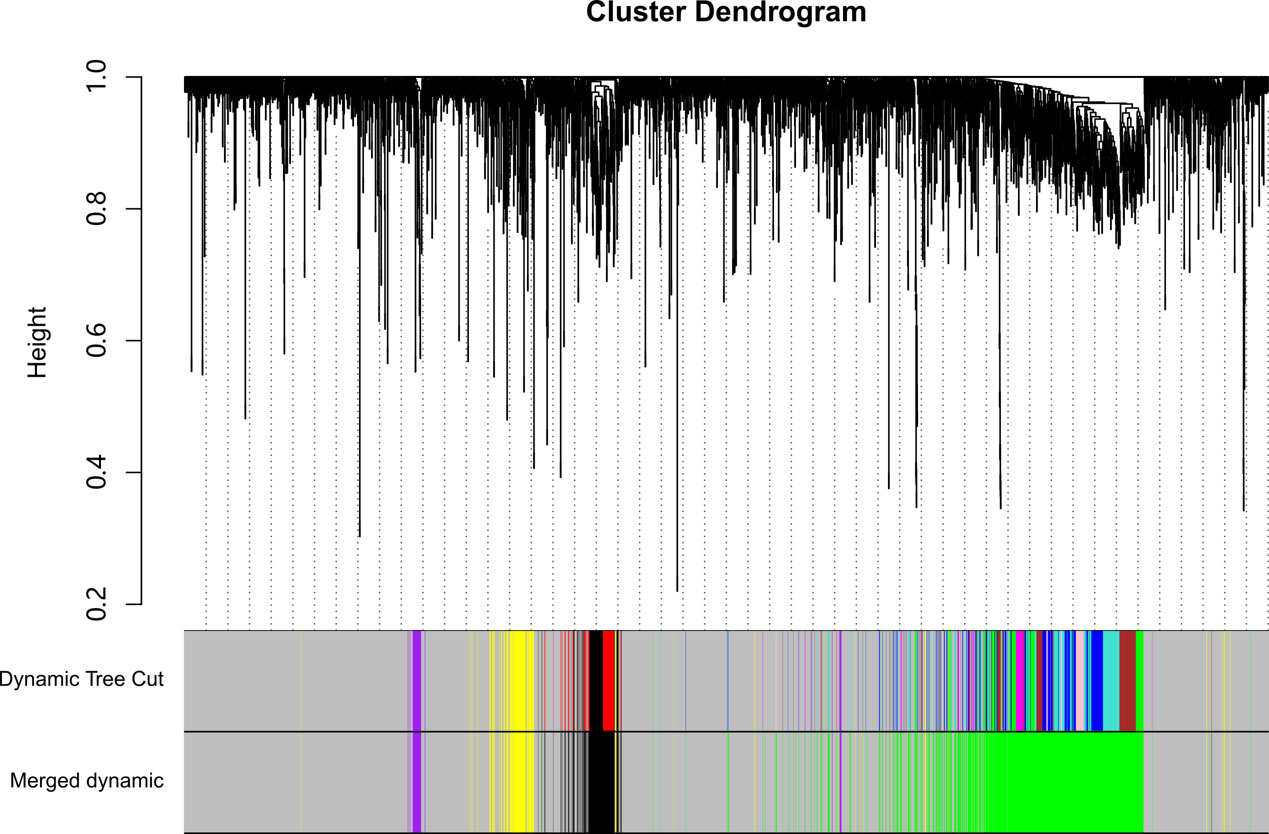
The relationship between modules and tissues. The left color labels on the left indicate the gene modules. The lower labels indicate the tissues of metastatic sites (ly: lymphoid; ad: adrenal; sk: skin; in: intestine; om: omentum). In the heatmap, red denotes a positive correlation and blue denotes a negative correlation between the eigengene and the tissue type vector. The two numbers in each heatmap blocks are the Pearson’s correlation coefficient and the 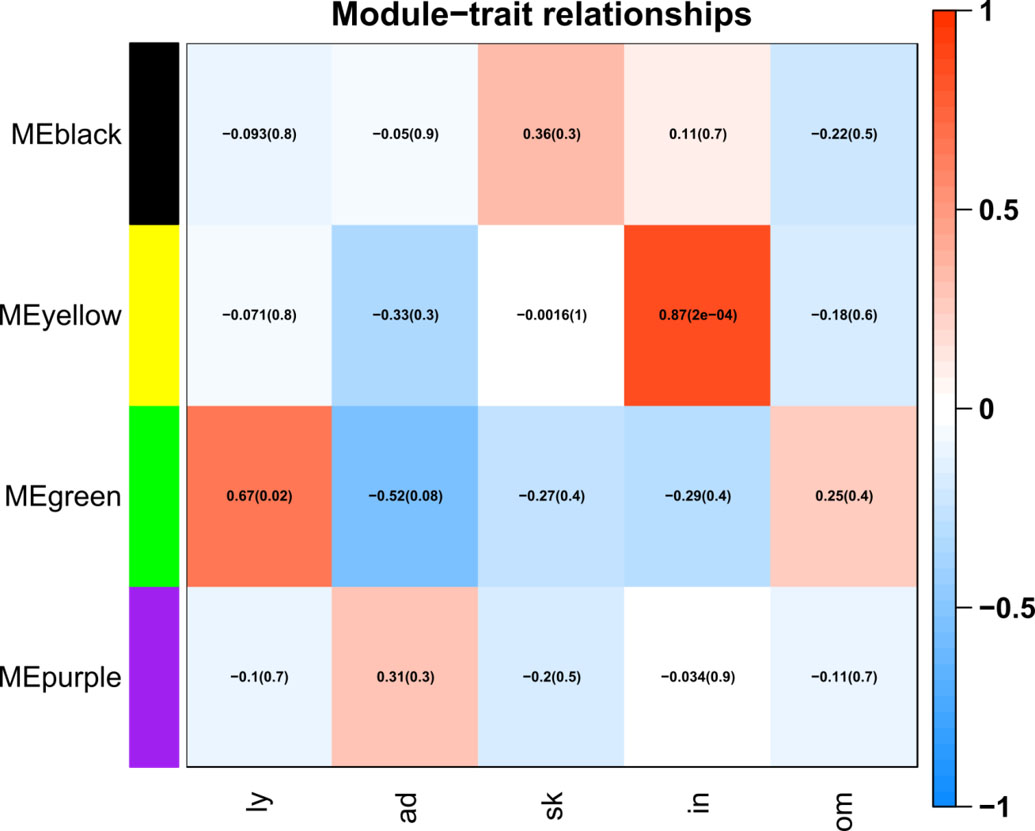
However, tumors are highly heterogeneous. It has long been realized that patients with the same metastatic site showed different survival time [4, 5]. Recently, the metastatic site was recognized as an important factor that affects clinical outcome [6, 7]. Riihimaki et al. [6] have found that liver and bone metastases caused a worse survival time, and the respiratory and nervous system metastases showed a better survival time. Gibson et al. [8] have found that liver metastasis had worse survival and adrenal metastasis had the best survival. Tamura et al. [9] have found that liver and adrenal gland metastasis had bad survival and brain and bone metastases have better survival. Those studies conflicted with each other for some metastatic sites. Commonly, liver metastasis had bad survival.
As for now, the understanding of the mechanism of the lethality difference within the metastases of lung cancer is still unclear. Only a limited number of studies in genomic variation analysis, especially in some driver genes, were reported. For examples, EGFR mutation could be an important marker for clinical outcome [10, 11]. The median survival time for the EGFR
In spite of the success in studying such driver genes, there are many unresolved questions. For examples, which mechanism is most important for the survival “Is there shared mechanism among these metastases” Is there a tissue-specific mechanism among these metastases?” In order to solve these problems, a global transcriptome study would be necessary.
In this study, the Weighted Correlation Network Analysis (WGCNA) was used to identify the gene modules in NSCLC. The tissue-specificity and function of those gene modules were studied. Further using an independant dataset, the clinical outcomes of these modules were evaluated. Specifically, some typical genes were carefully examined.
Prepare data for WGCNA analysis
By searching in the Gene Expression Omnibus (GEO) database (
WGCNA analysis for the gene modules
In order to get the co-expression gene modules, the WGCNA analysis was conducted with the 12 samples. As denoted by the analysis of the scale-free topology model fit, a soft power threshold at 37 showed a better performance. Unsigned Pearson’s correlation coefficient between genes was used to generate the adjacency matrix. The topological overlap was calculated to construct the co-expression network. Each gene was then hierarchically clustered according to the topological distance in the co-expression network (Fig. 2). The hierarchical tree was dynamically cut. Nine gene modules were inferred. The gene modules with high eigengene similarity (
Biological processes enriched in purple module. The 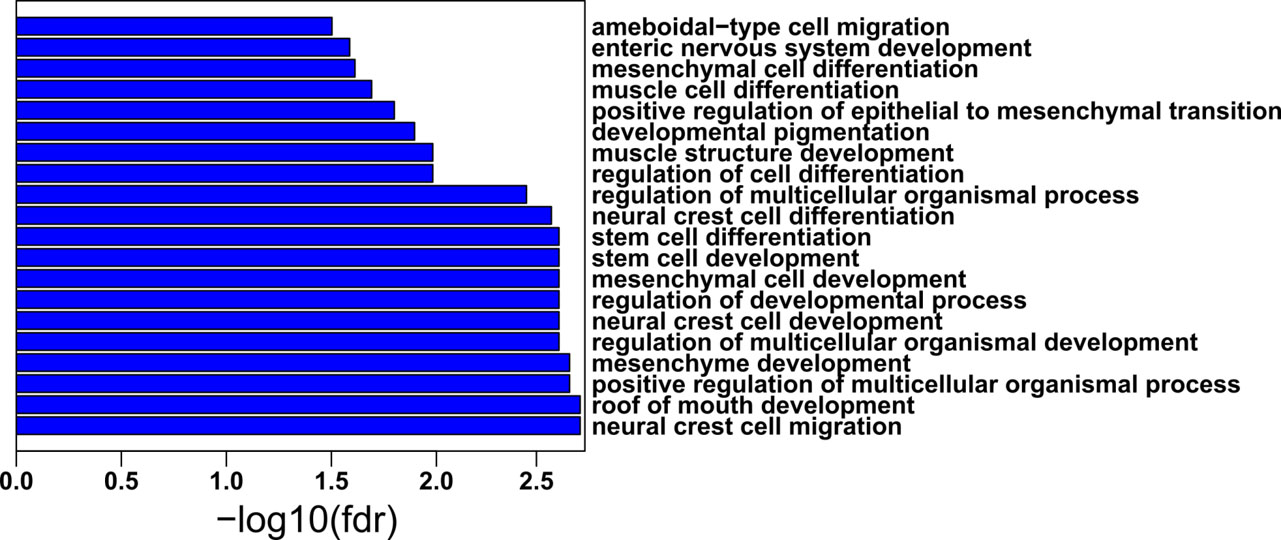
To characterize the gene modules, the eigengene was used as a surrogate measure for each gene module. A Pearson’s correlation coefficient between those eigengenes for each gene modules and the tissue type vector was calculated. As shown in (Fig. 3), the green module was highly expressed in the lymph node (
Study of module eigengenes by survival analysis
Study of module eigengenes by survival analysis
The study was conducted in a dataset consisting of 133 samples. The log-likelihood ratio test showed a significant correlation between the gene modules and the survival time. The green and purple gene modules were the two significant gene modules with hazard ratios, 0.45 and 2.76, respectively.
Next, the function of the four gene modules in the biological process was examined with gene ontology. The yellow module was enriched with “extracellular matrix organization” and “cell adhesion” (Suppl. Fig. 1A); the black module was enriched with “immune response”, “defense response”, “innate immune response” and “regulation of T cell activation” (Suppl. Fig. 1B); the green module was enriched with “hormone-mediated signaling pathway”, “cell communication” and “cellular response to endogenous stimulus” (Suppl. Fig. 1C); and the purple module was enriched with “neural crest cell migration”, “mesenchyme development” and “stem cell differentiation” (Fig. 4).
Except for the biological function of gene modules identified by WGCNA analysis, we also studied their clinical association. Without loss of generality, another dataset, GSE14814, was used to validate the gene modules. That dataset had recorded survival/censor time, which was used as a dependent variable in a Cox hazard proportional model (Table 1). The eigengene expressions of those gene modules was dichotomized by the median. They were used as independent variables in the model. As denoted by the log-likelihood ratio test, the model was significant with
Clinical significance of the typical genes from the purple module
Many studies [14, 15, 16] had shown that EMT was important for the metastasis process, but there were no studies about the clinical association with metastasis. In order to study the clinical significance of the EMT process, the hub genes and EMT related genes from the purple module were further validated in a wider dataset provided by the web service [17] (Fig. 5). The hub gene was defined as the gene which had the highest correlation with the eigengene of the gene modules. CA14 was the hub gene of the purple module. SOX10, ALX1, and TWIST1 were the genes involved in the EMT in the purple module. Using the median as the cutoff, their Kaplan-Meier curves were plotted as in Fig. 5. Higher expression of these genes showed a significantly shorter survival time with the
Validation of typical genes. CA14 is the hub gene of the purple module. SOX10, ALX1, and TWIST1 are three genes enriched in the EMT process. The four plots demonstrate the clinical significance of the four genes by Kaplan-Meier survival analysis. The Cox hazard proportional model and the log-rank tests gave the hazard ratio and 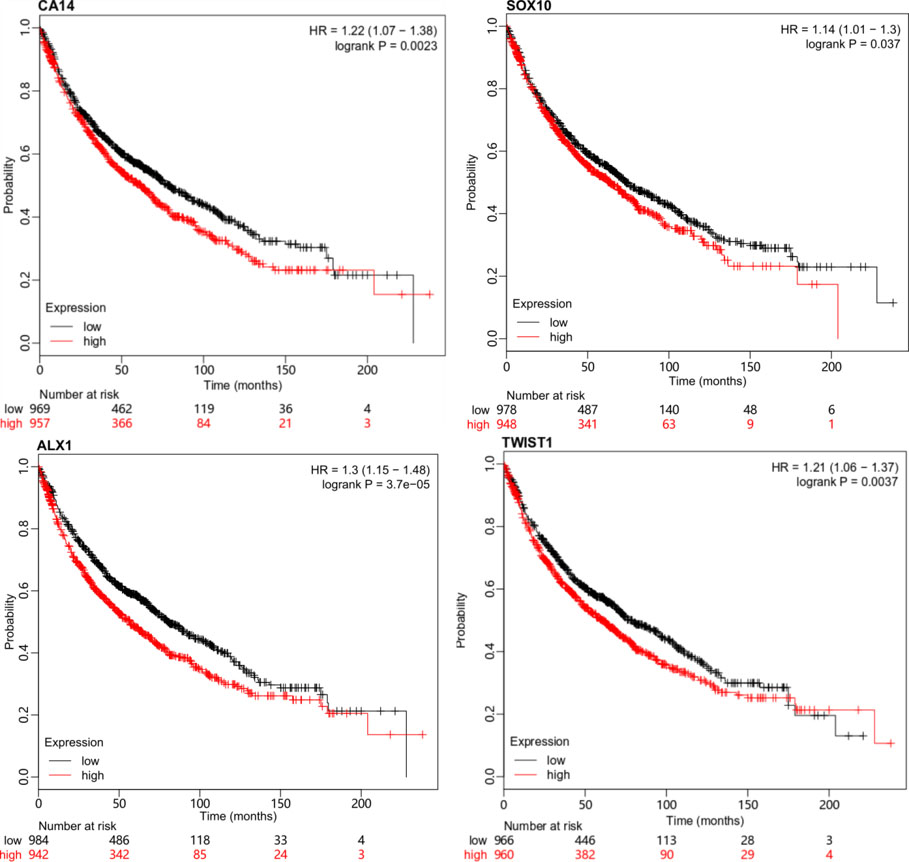
Lung metastasis could severely affect the clinical outcome [6]. Different metastatic sites may be correlated to significantly different survival [6, 8, 9, 10]. Those studies have found both common and different results. To resolve those discrepancies, it is urgent to determine the background molecular mechanisms. Compared to the genomic variations, the expression profile may reflect the biological process in those metastasis tissues more directly when considering the difficulties in the evaluation of the effects of genomic variations. Due to the difficult availability of metastatic tissues, there have been only a few studies on the expression profiles of lung cancer metastasis up to now. We search for relevant data in the GEO database (
After searching for the GEO database, only one dataset containing multiple metastatic tissues has been collected. That dataset only contains metastatic sites of the adrenal gland, lymphoid, skin and omentum tissues. There are no expression profiles from the liver, which is the most lethal metastasis site [6, 8, 9]. Limited by the data availability, it is hard to know whether the liver has a tissue-specific module or non-tissue-specific module. Brain metastasis is one of the most common metastatic sites of advanced lung cancer, and it is also one of the common metastatic sites leading to the death of lung cancer patients [18, 19, 20]. Although there is a study which contains only a single metastatic site of brain tissue [21], to avoid possible error from multiple experiments, it was not included in this study. Though the overall incidence of bone metastasis is unknown, bone metastasis is a frequent complication in patients with advanced cancer [22, 23]. However, there is also a lack of relevant data in this GEO data. We look forward to more efforts in this field of research.
In order to infer the gene modules that are tissue-specific and non-tissue-specific, unsupervised clustering is needed. In fact, in considering the high heterogeneity of lung cancer metastasis, Student’s t-test in combination with supervised clustering could always be futile. WGCNA has the advantage to cluster all genes into separate gene modules without the knowledge of the tissue types. In this study, we have successfully identified four gene modules. The function of those gene modules was studied with the tissue-specificity and the gene ontology of biological process. As we have hypothesized, two gene modules (green and yellow) are correlated with the tissue types and two other gene modules (black and purple) were not correlated with any tissue types.
The green module was correlated with lymphoid metastasis tissue. It was enriched with “cell communication”. The typical gene was Wnt10B, which was reported to be associated with lymph node metastasis of gastric cancer [24]. However, there is also a report in endometrial cancer that high expression of Wnt10B is prone to not have lymphoid metastasis [25]. Those results suggest that the expression of Wnt10B is critical for lymphoid metastasis. The green gene module could be tissue-specific for lymphoid metastasis of pan-cancer.
Similarly, the yellow module was correlated with intestine metastasis. It was enriched with “extracellular matrix organization” and “cell adhesion”. The typical genes were MMP2, COL1A1
The black gene module was non-tissue-specific and enriched with“immune response”, “defense response”, “innate immune response” and “regulation of T cell activation”. The black module reflects the function of immune microenvironment. It is a general mechanism in all types of cancers. For example, the inflammatory chemokine C–C motif ligand 4 (CCL4) is an “innate immune response”-related gene. CCL4 can promote the lymphangiogenesis in oral squamous cell carcinoma (OSCC), which is a critical step in tumor metastasis [29]. In breast cancer, CCL4 can contribute to the metastasis to the bone by binding to CC chemokine receptor type 5 [30]. The genes in the “regulation of T cell activation” category, HLA-DR or CD4, also play a critical role in the metastasis in the metastasis of melanoma [31], breast cancer [32], and prostate cancer [33].
The most interesting module was the purple module, which was also a non-tissue-specific gene module and had a significant effect on the survival. The purple module was enriched with“neural crest cell migration”, “mesenchyme development” and “stem cell differentiation”. Generally, for cancer metastasis, the nervous system plays a big part of metastatic initiation and clonal expansion, angiogenesis to extravasation and colonization [34]. The typical genes from it are SOX10, TWIST1
In summary, in this article, we have studied the gene co-expression modules in the metastatic tissues of lung cancer. Four gene modules were identified, from which two gene modules were tissue-specific and the other two gene modules were non-tissue-specific. Specifically, one of the non-tissue-specific gene modules was significantly correlated with survival. The function of that gene module demonstrates that the EMT process plays a big role in the metastasis process of lung cancer and survival.
Method
Data collections
Two datasets, GSE12630 [17] and GSE14814 [37], were downloaded from the Gene Expression Omnibus (GEO) website (
WGCNA analysis
WGCNA analysis was conducted on the R platform [38]. Briefly, the scale-free topology model fit was used to choose the soft power. An unsigned correlation distance defined as “
Gene function analysis
The biological processes of gene ontology were used to enrich the gene modules with the DAVID web service (
Survival analysis
The WGCNA modules were selected by survival analysis of their eigengenes. The eigengenes were dichotomized by the median. The expression over the median will be defined as high expression; the expression below the median will be defined as low expression. The gene modules were regressed against the survival time with Cox proportional hazards model with R package “survival” [40]. The log-likelihood test was used to test the significance. The modules with higher survival probability were further enriched with gene ontology. The typical genes were validated in a large dataset with the web service [17]. The median value was used as a cutoff to categorize the gene expression into high and low. The survival time was plotted against survival probability with the Kaplan-Meier curve. The log-likelihood ratio test was used to compare their different significance.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-201605.
sj-docx-1-cbm-10.3233_CBM-201605.docx - Supplemental material
Supplemental material, sj-docx-1-cbm-10.3233_CBM-201605.docx
Funding
This work was supported by the National Natural Science Foundation of China, grant nos. 81602009 (funder: Guanghui Wang) and 81672288 (funder: Jiajun Du) and The Joint Research Funds for Shandong University and Karolinska Institute, grant no. SDU-KI-2019-16 (funder: Jiajun Du).
Footnotes
Conflict of interest
Conflict of interest: Guanghui Wang, Fenglong Bie, Guangxu Li, Junping Shi, Yanwu Zeng and Jiajun Du declared no conflicts of interest in this work.