The aim of this study was to explore the mechanisms by which oral cancer acquires resistance to gemcitabine.
METHODS:
Oral squamous cell carcinoma (OSCC) cells were treated with gemcitabine upon infection or with a lentivirus harboring short hairpin RNA (shRNA) targeted to transforming growth factor- (TGF-). Then, Western blot, ELISA, migration assay, MTT assay, and animal experiments were used to explore the mechanism of resistance to gemcitabine treatment.
RESULTS:
After the treatment of non-transfected cells with gemcitabine, NF-B and AKT activities were increased, which may have induced the OSCC resistance to gemcitabine. Then, we found that TGF- downregulation effectively reduced NF-B and AKT phosphorylation levels after the administration of gemcitabine and increased the OSCC sensitivity to gemcitabine, resulting in cell death and the blunting of OSCC resistance to gemcitabine. The EMT was also reduced by TGF- downregulation combined with gemcitabine treatment.
CONCLUSION:
Cellular levels of TGF- constitute an important factor in gemcitabine resistance and TGF- silencing might represent a novel and potent strategy for overcoming OSCC resistance to gemcitabine.
Squamous cell carcinoma is the most commonly observed oral cancer in patients and represents approximately 1.9–3.5% of all cancers, being the second most prevalent head-and-neck cancer [1]. Moreover, its incidence is increasing at an alarming rate [2]. Current treatments include surgery, radiotherapy, and adjunctive chemotherapy but the effects of these treatments are not ideal, especially in the advanced stage when only chemotherapy could be selected, as resistance to chemotherapy results in low efficacy and poor prognosis.
Gemcitabine, a deoxycytidine nucleoside analog activated in cells by enzymatic reactions, inhibit DNA synthesis and induces apoptosis [3]. It can also inhibit multiple tumors, both in vitro and in vivo, as also confirmed in clinical trials [4]. Gemcitabine was described as a potential candidate for combination therapy in esophageal squamous cell carcinoma [5]. It can inhibit proliferation and induce apoptosis in OSCC cells. However, although Gemcitabine has certain effects on OSCC, it minimally affects the disease due to the resistance of cancer cells to the drug [6, 7, 8]. It is commonly known in the medical community that Akt and NF-B levels increase in cancer cells after the exposure to gemcitabine, indicating a possible mechanism of acquired resistance to this drug [9, 10, 11]. Therefore, molecules either enhancing gemcitabine’s effects or overcoming chemoresistance are required for efficient cancer therapy [12, 13].
TGF-, a major cytokine secreted in response to inflammatory stimuli, has a critical function in the development of multiple human malignant tumors, including oral carcinomas [14]. It is also important in the control of cell proliferation, angiogenesis, and tumor metastasis and is a critical component of the signaling network modulating tumorigenesis [15]. TGF-1 inhibits tumor cells and stops their growth in the early stage of carcinogenesis. In later disease stages, it induces epithelial to mesenchymal transition (EMT), cancer progression, angiogenesis, metastasis, and invasion [15]. Hino et al. reported in their previous study that TGF-1 induces OSCC cell invasion via Slug/Wnt-5b/MMP-10 signaling [16]. Previous studies have also shown that high expression of TGF- in oral squamous cell carcinoma is associated with immunosuppressive events that contribute to a worse prognosis in patients [7]. Considering that TGF- downregulation is sometimes sufficient in suppressing cancer cells, the combined application with gemcitabine may help in OSCC treatment.
Based on previous studies, we hypothesized that gemcitabine exerts anti-cancer effects in OSCC and that resistance to gemcitabine could be overcome by downregulation of TGF-. Thus, the current study aimed to explore the effects of gemcitabine in OSCC cells after TGF-downregulation in both in vitro and in vivo experiments to determine the efficacy and molecular target of the combined administration of TGF- and gemcitabine in human oral carcinoma cells and to provide suggestions for OSCC treatment in clinical applications.
Materials and methods
Cell lines and reagents
TCA8113 and Tb cells were maintained in DMEM containing 10% FBS at 37C in a humid environment with 5% CO.
Antibodies targeting phospho-Akt (Ser473), phospho-p65 (Ser536), and PARP were purchased from Cell Signaling (MA, USA). BCA Protein Assay was bought from Thermo (USA). All other reagents were bought from Sigma (USA).
The construction of retroviral vectors
The two micrograms of the resulting lentivirus transfer plasmids pCDH-shTGF-1/2 and pCDH together with 1.5 mg psPAX2 vector and 0.5 mg pMD2.G vector, respectively, were co-transfected into HEK-293T cells (Invitrogen) in a 10 cm plate and incubated for 72 hours at 37C and 5% CO2. Then, culture supernatants were centrifuged in Amicon Ultra-15 Centrifugal Filter Devices (3000 rpm, 30 minutes) for virus particle concentration.
TCA8113 and Tb cells were incubated in FBS-free medium as above for 48 hours. DMEM supplemented with 10 g/ml polybrene (Sigma) was refreshed daily. Next, 100 l of concentrated pCDH-shTGF-1/2 lentivirus was mixed with 1ml of the culture medium.
Cell viability assays
TCA8113 and Tb cells cultured in 96-well plates were administered in various doses of gemcitabine (0, 2.5, 5, 10, 15, and 20 M) for 24 hours and 48 hours, respectively. Cell viability after treatment of TGF- was also evaluated. The MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide) assay was employed to assess cell viability and expressed relatively to control the values.
Western blot
TCA8113 and Tb cells cultured in 6-well plates were administered with gemcitabine (IC50) for 24 hours and 48 hours, respectively and lysed. Protein amounts in the lysates were assessed with the BCA Protein Assay Kit. Anti-phospho-Akt (p-Akt), anti-p-p65), anti-PARP and anti-GAPDH primary antibodies were used for blotting.
Real-time RT-PCR
Cell lysis was performed with TRIzol reagent and total RNA was obtained via chloroform extraction. Real-time RT-PCR was carried out with the Power SYBR Green RNA-to-CT 1-Step Kit. Sense and antisense primers were synthesized based on a report for human TGF-1, TGF-2, and -actin mRNAs [17].
The measurement of the TGF-1 and TGF-2 levels by ELISA
TCa8113 and Tb cells cultured in 6-well plates were administered with gemcitabine (IC50) for 24 hours and 48 hours, respectively. Supernatants were obtained after 48 hours. Secreted TGF-1/2 amounts were determined by ELISA using specific kits from R&D Systems, as instructed by the manufacturer.
NF-B activity assessment
TCa8113 and Tb cells were plated in 48-well plates, followed by transfection with a detection plasmid from the Cignal Reporter Assay kit (QIAGEN) after 24 hours. Each cell line was treated with experimental conditions the following day. Luciferase activity was assessed based on the Dual-Luciferase Reporter Assay System Protocol (Promega).
Clonogenic assays
Cells plated in 6-well plates were treated with experimental conditions of gemcitabine or gemcitabine combined with the lentivirus for 24 hours and 48 hours, respectively and re-plated in 6-well plates at the same density. Upon the colony formation, fixation with 4% formalin was performed followed by staining with a 0.5% crystal violet.
Transwell migration assays
Cells cultured in 10 cm dishes were administered with the experimental conditions of gemcitabine or gemcitabine combined with the lentivirus for 48 hours. The migration assay was carried out with a Transwell system containing 8 m-pore inserts (BD Biosciences, USA) according to the manufacturer’s instructions.
Animal studies
TCa8113 cells (1 10) in 100 L of PBS solution were administered to BALB/c nude mice subcutaneously in the abdominal region. The mice were randomly assigned to 5 groups ( 8), including the PBS, gemcitabine, NCgemcitabine, shT1gemcitabine, and shT2gemcitabine groups. After the tumor volumes approximated 70–100 mm, the intratumoral administration of PBS or 2 10 virus particles (VP) of lentiviruses in 50 l PBS was performed. This treatment was repeated three times and 50 l of gemcitabine (10 M) was co-administered on the day of the final lentiviral administration. After 7 days of various treatments, the effects on tumor growth and survival rates were measured and recorded.
Immunohistochemistry
Seven days after the final lentiviral administration, the tumor tissue samples were obtained and submitted to 10% formaldehyde fixation for more than 24 hours. IHC was carried out with anti-TGF-1 (Abcam), anti-TGF-2 (Abcam), and anti-ki67 primary antibodies. After deparaffinization with xylene and rehydration with an ethanol gradient, endogenous peroxidase was quenched by treatment with 0.1% HO. Then, a 10 mM citrate buffer (pH 6.0) was employed for antigen retrieval. Upon permeabilization (0.5% PBX) and blocking (5% BSA), the samples were incubated with primary antibodies overnight. HRP-conjugated secondary antibodies (Thermo Scientific) were employed for detection. Finally, DAB Plus Chromogen and DAB Plus Substrate (Thermo Fisher Scientific) were used for the color display.
Statistical analysis
Statistical analyses were performed with Graph Pad (Systat Software, USA). A -value of 0.05 indicated statistical significance.
Results
Gemcitabine induces changes in the survival and death-associated signaling pathways in OSCC
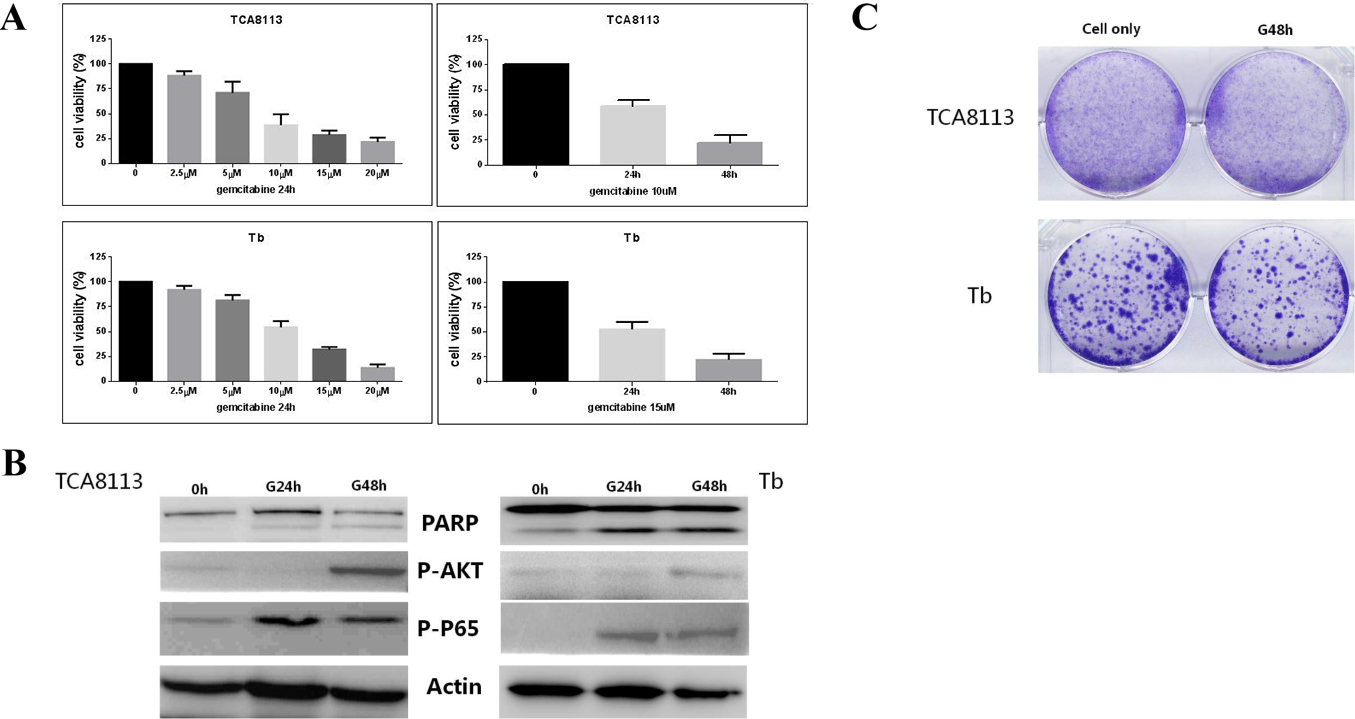
To explore the effects of gemcitabine in the cell cultures, we firstly determined the IC50 values of gemcitabine in various OSCC cell lines, which ranged between 2.5 M and 20 M (Fig. 1A). Then, key effectors in the survival and death signaling pathways were assessed, including PARP, p-p65, and p-AKT by immunoblot after the treatment with gemcitabine in the OSCC cell lines. The results showed that the phosphorylation levels of AKT and P65 were increased by the gemcitabine treatment while the cleaved form of PARP was not increased in a time-dependent manner (Fig. 1B). We also evaluated the cell viability after the treatment with gemcitabine by clonogenic assay and cell survival was high, even after the treatment with gemcitabine. These findings indicated the low effect of gemcitabine on OSCC, as well as the resistance of OSCC to gemcitabine treatment (Fig. 1C). Elevated p-p65 and p-Akt (Fig. 1B) levels suggested that the OSCC cells were gemcitabine resistant.
The effect of gemcitabine in different oral carcinoma cell lines. A: IC50 values for gemcitabine in various OSCC cell lines. B: Western blot of PARP, P-AKT, and P-P65. Concentration of gemcitabine 15 M. C: Clonogenic assay of OSCC. Concentration of gemcitabine 15 M.
Changes in the TGF- expression detected by ELISA in response to the gemcitabine treatment in oral carcinoma cell lines. A: The expression of TGF-1 and TGF-2 after different incubation times of gemcitabine. B: The expression of TGF-1 and TGF-2 after incubation times of gemcitabine in gemcitabine-resistant cells. C: Cell viability after the treatment with the TGF- protein. D: The expression of TGF-1 and TGF-2 after the incubation of shRNA of TGF-1/2.
The TGF- expression is increased by gemcitabine treatment in OSCC
After the administration of gemcitabine, the TGF- levels in cell supernatants were determined by ELISA. As shown in Fig. 2A, gemcitabine increased the TGF-1 expression in a time-dependent manner. TGF-1 and TGF-2 expressions were also dramatically elevated in gemcitabine resistant cells, which were the cells that were still alive after incubation of gemcitabine at IC50 (Fig. 2B).
TGF- expression in oral carcinoma cell lines reduces gemcitabine sensitivity. A: Cell viability after the incubation of shRNA of TGF-1/2 and gemcitabine. B: NF-kB activity after the incubation of shRNA of TGF-1/2 and gemcitabine. C: Western blot of PARP, P-AKT, and P-P65 after the incubation of shRNA of TGF-1/2 and gemcitabine. D: Clonogenic assay after the incubation of shRNA of TGF-1/2 and gemcitabine.
Cell viability was also significantly reduced by the treatment with the TGF- protein (Fig. 2C). We designed a lentivirus harboring the shRNA of TGF-1/2, which can downregulate TGF-1/2 (Fig. 2D).
The co-treatment of gemcitabine after lentivirus infection is more effective in killing OSCC cells
As shown in Fig. 3A, cell viability was effectively reduced in the combination group compared with the NC virus-infected group. NF-kB activity was also decreased dramatically in the combination treatment group but increased by gemcitabine when used alone (Fig. 3B). Subsequently, we screened various signaling pathway molecules, including PARP, p65, and AKT. We observed that the p-AKT and p-P65 amounts were elevated after treatment with gemcitabine alone but decreased in the combination group; meanwhile, cleaved PARP levels were effectively increased (Fig. 3C). The emergence of clones originated from TCA8113 was reduced dramatically in the combination group (Fig. 3D).
The co-treatment of gemcitabine and the lentivirus-expressing shRNA against TGF- could suppress EMT and migration
The EMT includes physiological and pathophysiological events in which epithelial cells are converted into mesenchymal cells for functional needs. In cancer, the EMT contributes to the early-stage dissemination of cancer cells followed by invasion and metastasis, as well as drug resistance.
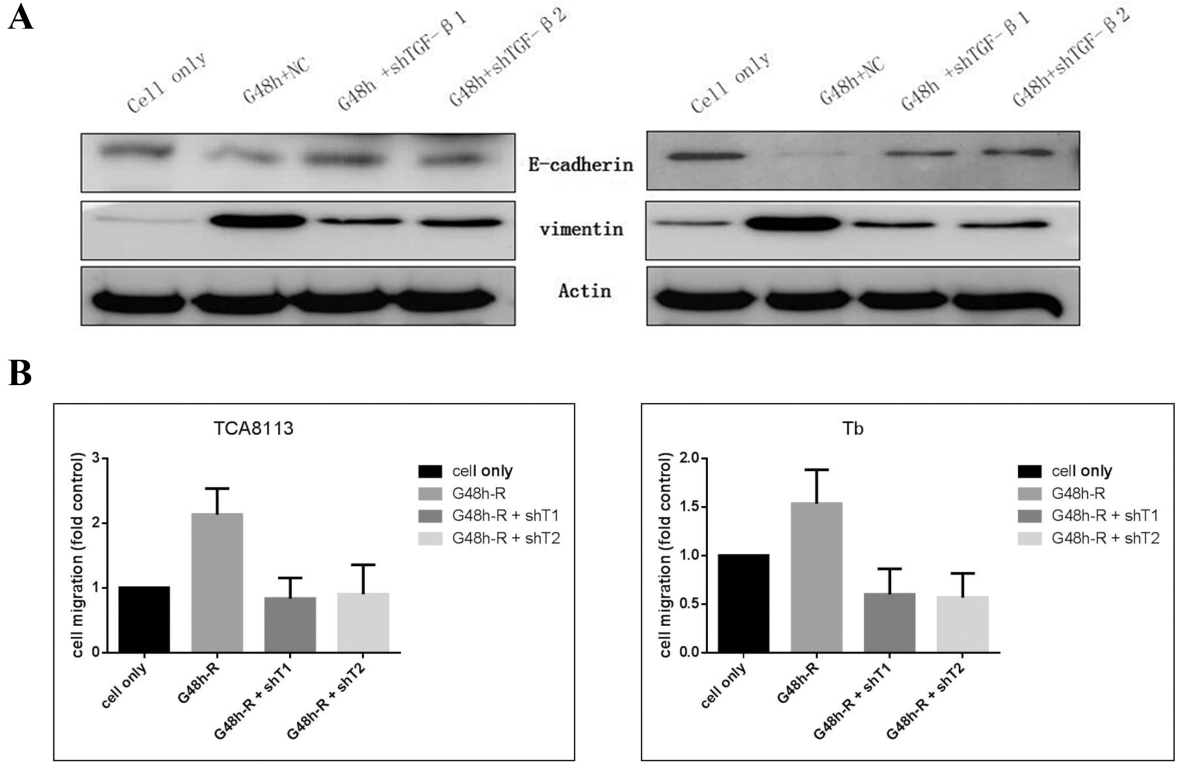
As shown in Fig. 4A, the exposure of TCA8113 and Tb cells to gemcitabine caused the upregulation of the mesenchymal marker vimentin while markedly decreasing epithelial marker E-cadherin levels. However, the gemcitabine-induced regulation of EMT markers was impaired by pretreatment with the lentivirus downregulating the expression of TGF-.
We reduced the expression of TGF- reprograms resistance-induced epithelial to mesenchymal transition and migratory behavior. A: Western blot of EMT markers after the incubation of shRNA of TGF-1/2 and gemcitabine. B: Transwell migration assays after the incubation of shRNA of TGF-1/2 and gemcitabine.
Moreover, Transwell migration assays demonstrated that the amounts of migrated cells after TGF- downregulation were reduced in comparison with the gemcitabine resistance groups ( 0.001) (Fig. 4B).
In vivo anti-tumor effects of the co-treatment of gemcitabine and the lentivirus expressing shRNA against TGF-
Since the abovementioned lentivirus expressing shTGF-1/2 enhanced apoptosis and decreased TGF-1/2 levels, thereby overcoming TCA8113 cell resistance to gemcitabine, we designed an in vivo experiment to further assess the anti-tumor effects of this therapeutic combination in xenograft animal models. Whether the combination could overcome TCA8113 cell resistance to gemcitabine was determined.
The results showed that treatment with gemcitabine followed by the lentivirus expressing shTGF-1/2 exerted enhanced anti-tumor effects in comparison with gemcitabine as monotherapy (Fig. 5A). However, the resistance was not entirely abolished. Animal survival was highest after administering the combined therapy with the lentivirus expressing shTGF-1/2 and gemcitabine with no mice dying until 19 days post-treatment (Fig. 5B). At this point, the gemcitabine alone and PBS groups displayed survival rates of 40% and 0%, respectively (Fig. 5B).
The anti-tumor effects of the combined treatment of gemcitabine and Lentivirus expressing shTGF-1/2 in BALB/c nude mice. A: The tumor size after the treatment of shRNA of TGF-1/2 and gemcitabine. B: The survival rate after the treatment of shRNA of TGF-1/2 and gemcitabine. C: The expression of Ki-67, TGF-1, and TGF-2 after the treatment of shRNA of TGF-1/2 and gemcitabine.
The expression levels of Ki-67 were also markedly elevated in the gemcitabine alone group and reduced in the combination group. The staining index of Ki-67 after administering the combined treatment was significantly reduced in comparison to the resistance group.
As shown in the ex vivo experiments, combined treatment with the lentivirus expressing shTGF-1/2 and gemcitabine noticeably increased the anti-tumor effects while effectively overriding gemcitabine resistance in animal models of oral cancer.
Discussion
Gemcitabine can inhibit many cancers, e.g. breast, head-and-neck, bladder, ovary, lung, and pancreatic cancers [18, 19, 20]. One of its well-known anticancer mechanisms is its interference with DNA replication and inhibition of DNA synthesis [21, 22, 23]. However, its inhibitory effects on OSCC is not very ideal. Previous studies have shown that the high expression of TGF- in OSCC is associated with immunosuppressive events that contribute to a worse prognosis in patients. Therefore, in the current study, we explored the effects of gemcitabine in combination with gene therapy in OSCC.
Firstly, we found that gemcitabine at IC50 (10 M) could not induce the activation of PARP cleavage and the cell survival rate was still very high after the administration of a low dose of gemcitabine (Fig. 1). p-Akt and p-p65 activities were also enhanced by low-dose gemcitabine (Fig. 1). Therefore, we hypothesized that the potential mechanism of resistance might be decreased by PARP cleavage. Increased cell death signals, e.g. by gemcitabine and shTGF- treatment, eventually resulted in reduced NF-B and p-Akt activities. These results suggest that the enhanced cell viability after gemcitabine (10 M) administration is associated with drug resistance via increased TGF- expression. However, reduced cell viability after TGF- administration is not associated with drug resistance. Based on the above results, we reduced TGF- by using a lentivirus expressing shRNA against TGF- to enhance gemcitabine cytotoxicity while reducing resistance. NF-B activity was obviously decreased after treatment with the combination of gemcitabine and lentivirus-shTGF-. The different death-inducing agents could improve cell death while finally resulting in decreased NF-B activity (Figs 2 and 3).
The elevated expression of TGF- in patients with advanced cancer promotes tumor growth, metastasis, angiogenesis, and EMT and inhibits the anti-tumor immune response [24, 25, 26]. Therefore, we assessed vimentin and E-cadherin in oral carcinoma cells (Fig. 4). The results showed that the E-cadherin levels in these cells were negatively correlated with the treatment with gemcitabine. The higher the expression of vimentin, the more pronounced the drug resistance. However, these effects were reversed by TGF- downregulation. We also confirmed that the combined use of shTGF- and gemcitabine resulted in decreased cell migration, reduced TGF- expression, and inhibited EMT progression while effectively overcoming gemcitabine drug resistance.
Subsequently, the effects of the combination of gemcitabine and shTGF- to overcome drug resistance were assessed in live animals. Our results revealed that the combined shTGF-1/2 and gemcitabine had the most substantial anti-tumor effects and effectively overcame OSCC cell resistance to gemcitabine. However, there were no significant differences between the shTGF-1 and shTGF-2 groups. This may result from the differences in the expression levels of target proteins in tumors or viral residence time. We also demonstrated that the Ki-67 amounts were elevated in gemcitabine-treated cells but reduced in the combination treatment group. In this study, we used lentiviruses, which cannot replicate in tumor cells, to assess the role of shRNAs in overcoming drug resistance. Supplementary studies using oncolytic viruses in immune-competent animal models, which closely reflect the clinical condition, should be carried out.
As the above mentioned animal study showed that tumor regression was not complete after the combination treatment, more efficient ways to improve anti-tumor effects and overcome chemoresistance are needed. In future studies, immune-competent animal models should be assessed to provide more generalizable findings.
Conclusion
In this study, we found that PARP cleavage inhibition was involved in chemoresistance of gemcitabine and that TGF- downregulation increased the cytotoxicity of gemcitabine and decreases OSCC cell resistance to this drug.
Footnotes
Acknowledgments
We are particularly grateful to all the people who have given us help on our article. The study was supported by a grant from the National Natural Science Foundation of China (No.81560179).
Conflict of interest
The authors declare that they have no competing interests.
References
1.
KademaniD., Oral cancer, Mayo Clinic Proceedings82(7) (2007), 878–887.
2.
ChenS.W. et al., Transfection of the nm23-H1 gene into BcaCD885 cell line inhibits the potential of invasion, adhesion and mobility, Zhonghua Kou Qiang Yi Xue Za Zhi = Zhonghua Kouqiang Yixue Zazhi = Chinese Journal of Stomatology38(1) (2003), 16–19.
3.
NakashimaM. et al., Phosphorylation status of heat shock protein 27 plays a key role in gemcitabine-induced apoptosis of pancreatic cancer cells, Cancer Letters313(2) (2011), 218–225.
4.
PlunkettW. et al., Gemcitabine: Preclinical pharmacology and mechanisms of action, Seminars in Oncology23(5 Suppl 10) (1996), 3–15.
5.
MynhardtC. et al., Metformin-induced alterations in nucleotide metabolism cause 5-fluorouracil resistance but gemcitabine susceptibility in oesophageal squamous cell carcinoma, Journal of cellular biochemistry119(1) (2018), 1193–1203.
6.
HilbigA. and OettleH., Gemcitabine in the treatment of metastatic pancreatic cancer, Expert Review of Anticancer Therapy8(4) (2008), 511–523.
7.
BafnaS. et al., Pancreatic cancer cells resistance to gemcitabine: The role of MUC4 mucin, British Journal of Cancer101(7) (2009), 1155–1161.
8.
DuxburyM.S. et al., Inhibition of SRC tyrosine kinase impairs inherent and acquired gemcitabine resistance in human pancreatic adenocarcinoma cells, Clinical Cancer Research: An Official Journal of the American Association for Cancer Research10(7) (2004), 2307–2318.
9.
TabaK. et al., Heat-shock protein 27 is phosphorylated in gemcitabine-resistant pancreatic cancer cells, Anticancer Research30(7) (2010), 2539–2543.
10.
ArltA. et al., Role of NF-kappaB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine-induced cell death, Oncogene22(21) (2003), 3243–3251.
11.
BanerjeeS. et al., Molecular evidence for increased antitumor activity of gemcitabine by genistein in vitro and in vivo using an orthotopic model of pancreatic cancer, Cancer Research65(19) (2005), 9064–9072.
12.
ChenD. et al., Inhibition of AKT2 enhances sensitivity to gemcitabine via regulating PUMA and NF-κB signaling pathway in human pancreatic ductal adenocarcinoma, International Journal of Molecular Sciences13(1) (2012), 1186–1208.
13.
HilbigA. and OettleH., Gemcitabine in the treatment of metastatic pancreatic cancer, Expert Review of Anticancer Therapy8(4) (2008), 511–523.
14.
KunnumakkaraA.B. et al., Curcumin potentiates antitumor activity of gemcitabine in an orthotopic model of pancreatic cancer through suppression of proliferation, angiogenesis, and inhibition of nuclear factor-kappaB-regulated gene products, Cancer Research67(8) (2007), 3853–3861.
15.
ChengC.M. et al., Up-regulation of miR-455-5p by the TGF-β-SMAD signalling axis promotes the proliferation of oral squamous cancer cells by targeting UBE2B, The Journal of Pathology240(1) (2016), 38–49.
16.
HinoM. et al., Transforming growth factor-β1 induces invasion ability of HSC-4 human oral squamous cell carcinoma cells through the Slug/Wnt-5b/MMP-10 signalling axis, Journal of Biochemistry159(6) (2016), 631–640.
17.
KangD.X. et al., Down-regulation of TGF-β expression sensitizes the resistance of hepatocellular carcinoma cells to sorafenib, Yonsei Medical Journal58(5) (2017), 899–909.
18.
SpielmannM. et al., Single-agent gemcitabine is active in previously treated metastatic breast cancer, Oncology60(4) (2001), 303–307.
19.
BraakhuisB.J. et al., Preclinical in vivo activity of 2’,2’-difluorodeoxycytidine (Gemcitabine) against human head and neck cancer, Cancer Research51(1) (1991), 211–214.
20.
CarmichaelJ., The role of gemcitabine in the treatment of other tumours, British Journal of Cancer78(Suppl 3) (1998), 21–25.
21.
PlunkettW. et al., Gemcitabine: Metabolism, mechanisms of action, and self-potentiation, Seminars in Oncology22(4 Suppl 11) (1995), 3–10.
22.
UenoH.KiyosawaK. and KaniwaN., Pharmacogenomics of gemcitabine: Can genetic studies lead to tailor-made therapy? British Journal of Cancer97(20) (2007), 145–151.
23.
PauwelsB. et al., Combined modality therapy of gemcitabine and radiation, The Oncologist10(1) (2005), 34–51.
24.
MassaguéJ., TGFbeta in cancer, Cell134(2) (2008), 215–230.
25.
LinT.H. et al., High serum transforming growth factor-β1 levels predict outcome in hepatocellular carcinoma patients treated with sorafenib, Clinical Cancer Research: An Official Journal of the American Association for Cancer Research21(16) (2015), 3678–3684.
26.
TanQ.Y. and ChengZ.S., TGFβ1-smad signaling pathway participates in interleukin-33 induced epithelial-to-mesenchymal transition of A549 cells, Cellular Physiology and Biochemistry: International Journal of Experimental Cellular Physiology, Biochemistry, and Pharmacology50(2) (2018), 757–767.