
Abstract
Previous studies have suggested potential signature genes for lung cancer, however, due to factors such as sequencing platform, control, data selection and filtration conditions, the results of lung cancer-related gene expression analysis are quite different. Here, we performed a meta-analysis on existing lung cancer gene expression results to identify Meta-signature genes without noise. In this study, functional enrichment, protein-protein interaction network, the DAVID, String, TfactS, and transcription factor binding were performed based on the gene expression profiles of lung adenocarcinoma and non-small cell lung cancer deposited in the GEO database. As a result, a total of 574 differentially expressed genes (DEGs) affecting the pathogenesis of lung cancer were identified (207 up-regulated expression and 367 down-regulated expression in lung cancer tissues). A total of 5,093 interactions existed among the 507 (88.3%) proteins, and 10 Meta-signatures were identified: AURKA, CCNB1, KIF11, CCNA2, TOP2A, CENPF, KIF2C, TPX2, HMMR, and MAD2L1. The potential biological functions of Meta-signature DEGs were revealed. In summary, this study identified key genes involved in the process of lung cancer. Our results would help the developing of novel biomarkers for lung cancer.
Introduction
Lung cancer, which accounts for the most percentage of morbidity and mortality from cancer, injures people all over the world [1, 2, 3, 4, 5]. It is reported that only in 2018, there were about 2.1 million incidences and 1.8 million deaths [6, 7]. In theory, lung cancer can be classified into two categories, which are small cell lung cancer (SCLC) and non-small cell lung carcinoma (NSCLC) [7]. Worse still, lung cancer is usually diagnosed at a later stage of the disease, which limits its possibility for treatment [8]. Although many efforts have been done for lung cancer, such as surgical therapy, chemotherapy, and molecular targeting therapy, the prognosis of lung cancer and the 5-year survival rate are still very low [9].
Lung cancer is diagnosed at the molecular level, so it is needed to figure out the biological mechanisms of it to find out effective therapies [10]. It is reported that lung cancer contains various cell clones, corresponding to their only molecular signature, respectively [10, 11]. The therapies of lung cancer are different among patients because of the presence of genetic variations in the patient’s tumor [12].
In this study, we intended to achieve the following two goals: one is to provide reliable genetic information related to lung cancer; the other is to analyze the biological functions of differentially expressed genes in the pathogenesis of lung cancer, and provide a theoretical basis for the use of related genes as a biomarker to prevent and treat lung cancer.
Analysis process for this study.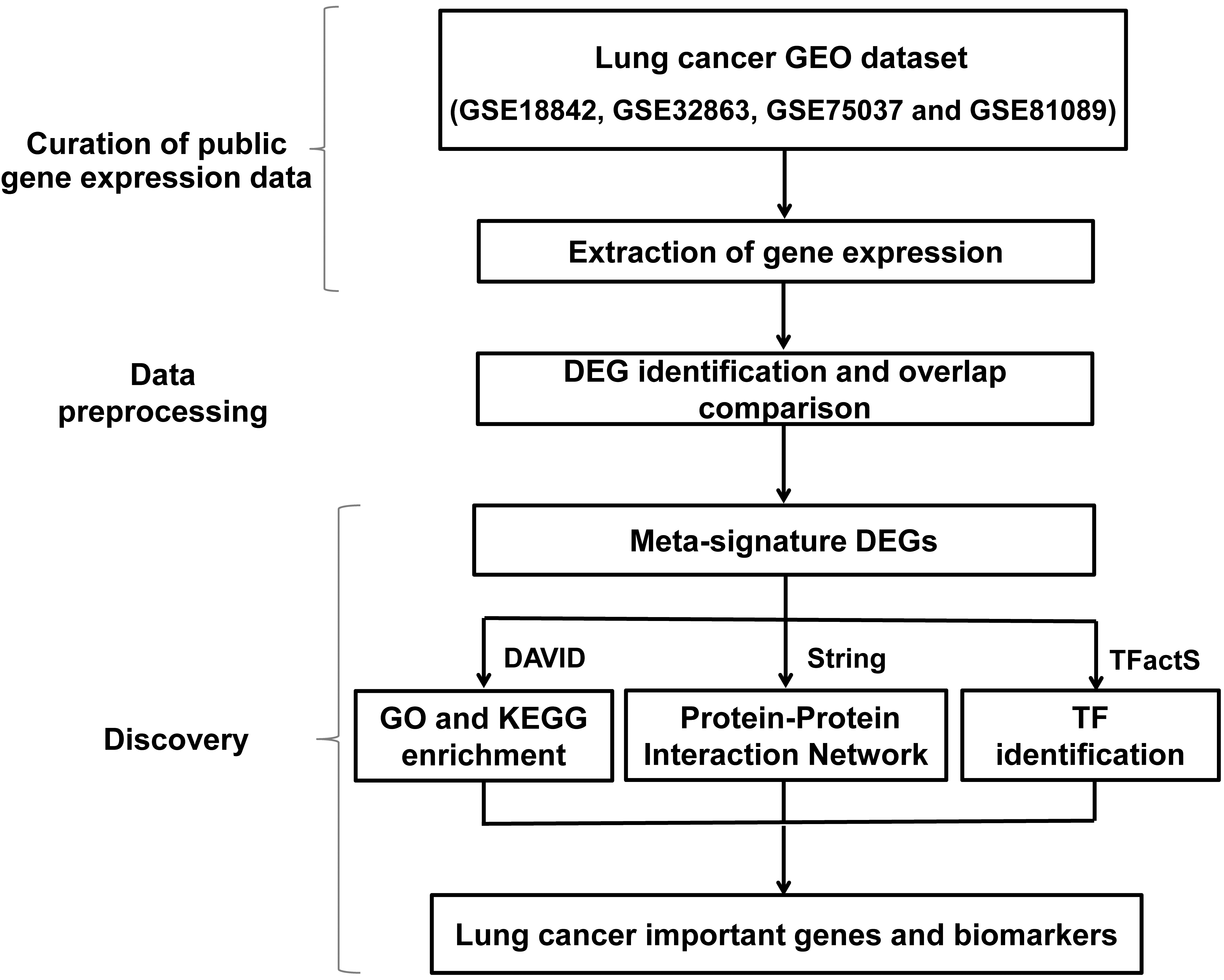
Identification of lung cancer gene expression data sets and differentially expressed DEGs
NCBI’s GEO database (
Functional enrichment analysis of differential expression of meta-signature DEGs in lung cancer
The up-and-down DEGs were mapped to the DAVID database [13]. The Gene Ontology (GO) [14] annotation and KEGG Pathway [15, 16] were further analyzed. The filter condition was that the
Protein-protein interaction network construction and analysis for Meta-signature DEGs in lung cancer
The lung cancer differential expression Meta- signature DEGs were mapped to the String database (
Transcription factor analysis of differentially expressed Meta-signature DEGs in lung cancer
The lung cancer differential expression Meta- signature DEGs were submitted to the Tfacts database (
Results
In this study, the existing lung cancer-related GEO data (GSE18842, GSE32863, GSE75037, and GSE81089) was applied to explore important genes affecting the pathogenesis of lung cancer, determine the relevant biomarkers for lung cancer, and explore the possible mechanisms of genes involved in lung cancer [21, 22, 23, 24]. The key up- and down-regulation genes of lung cancer tissues compared with the control were determined by meta-analysis [25, 26]. The functional enrichment, protein-protein interaction network and transcription factor binding analysis of these DEGs were further analyzed. The work flow for the analysis was shown in Fig. 1.
First, the DEGs was determined by comparing the existing lung cancer gene expression results from multiple datasets. Functional enrichment analysis, protein-protein interaction network analysis, and transcription factor binding analysis of up and down DEGs were used to investigate the possible mechanisms of these genes affecting the pathogenesis of lung cancer and to study their biological functions.
Identification of differentially expressed genes related to lung cancer
Based on “foldchange
Distribution of differentially expressed genes in four lung cancer datasets. A. The lung cancer dataset focuses on the up-regulation of the number of genes expressed. A total of 6222 up-regulated genes were obtained, and only 12 genes were up-regulated in all four data sets. Select genes that are up-regulated in at least 3 data as Meta-signature genes, totaling 207. B. The lung cancer dataset down-regulates the quantitative statistics of expressed genes. A total of 3621 genes down-regulated were obtained, and only 49 genes were down-regulated in all four data sets. Genes up-regulated in at least 3 data were selected as Meta-signature genes, for a total of 367.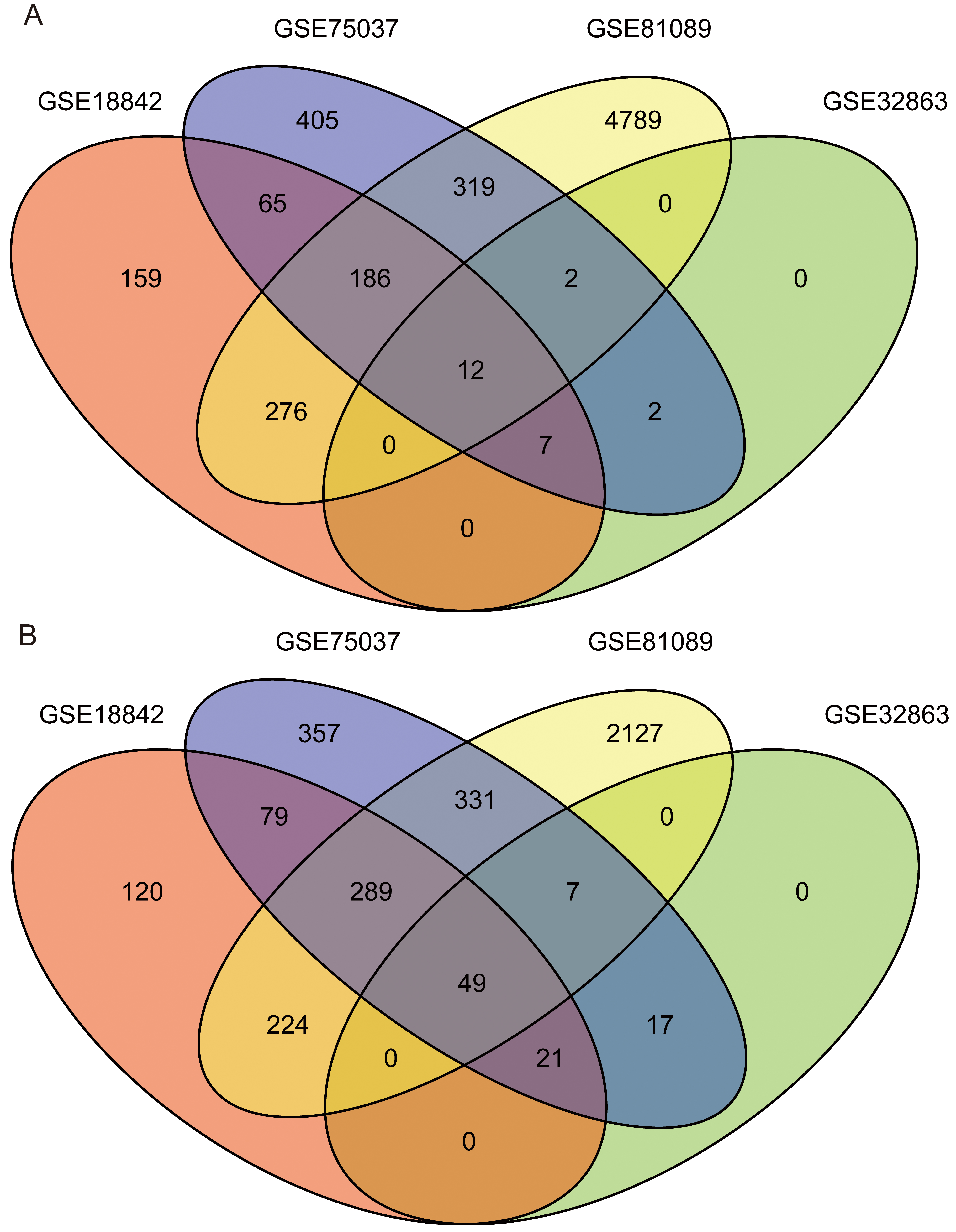
GO enrichment analysis was performed on up and down DEGs. The results showed that the up-regulated expression of Meta-signature genes was mainly involved in mitotic nuclear division (GO: 0007067), cell division (GO: 0051301), sister chromatid condensation (GO: 0007062), chromosome separation (GO: 0007059), G1/S transition of mitotic cell cycle (GO: 000082) and other biological processes (Fig. 3A). While down-regulated expression of Meta-signature genes were mainly involved in angiogenesis (GO: 0001525), leukocyte migration (GO: 0050900), cell adhesion (GO: 0007155), immune response (GO: 0006955), vasculogenesis (GO: 0001570
Basic information on lung cancer gene expression data sets
Basic information on lung cancer gene expression data sets
Up- and down-regulation of differentially expressed genes in GO biological processes and KEGG metabolic pathways enrichment results. A. Up-regulation of differentially expressed genes GO biological process enrichment results. B. Down-regulation of differential expression target gene GO biological process enrichment results. C. Up-regulation of differentially expressed genes KEGG metabolic pathway enrichment results. D. Down-regulation of differentially expressed genes KEGG metabolic pathway enrichment results.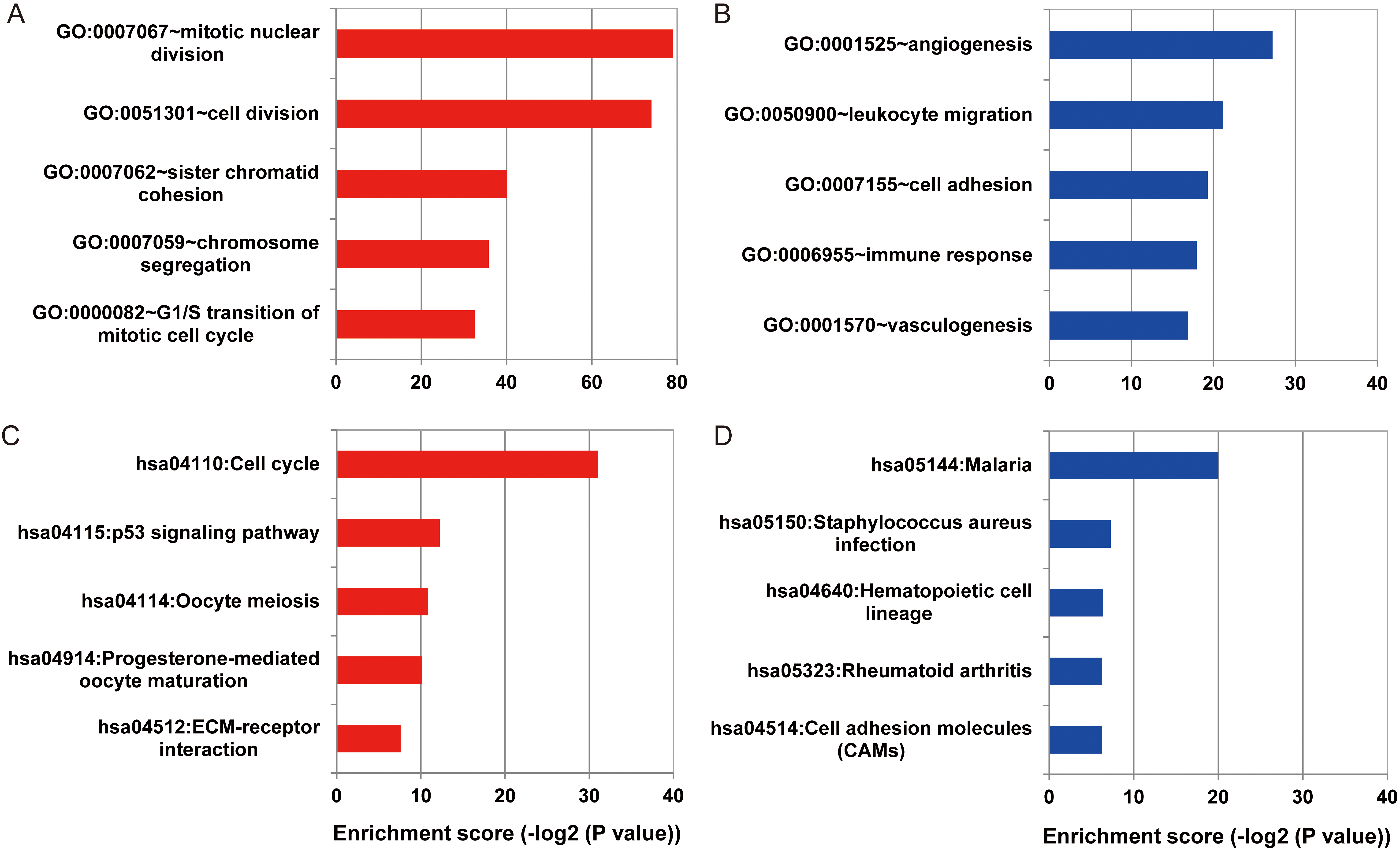
The results of KEGG enrichment showed that the up-regulated expression of Meta-signature genes were mainly enriched in the cell cycle (hsa04110), p53 (mutated) signaling pathway (hsa04115), oocyte meiosis (hsa04114), progesterone-mediated oocyte maturation and ECM-receptor interaction (hsa04512) and other metabolic pathways (Fig. 3C). While down-regulated expression of the meta-signature genes were mainly enriched in malaria (hsa05144), Hematopoietic cell lineage (hsa05150), rheumatoid arthritis (hsa05323), cell adhesion molecules (hsa04514) and other metabolic pathways (Fig. 3D).
The interaction between the corresponding proteins of Meta-signature Genes was analyzed using String database and Cytoscape software. As shown in Fig. 4, a total of 5,093 interactions were present among 507 (88.3%) of the total 574 proteins.
Furthermore, the number of hub genes in the above protein-protein interaction network was calculated and the genes corresponding to the top 10 proteins were extracted (Table 2). The results showed that the strongest hub genes were AURKA, CCNB1, KIF11, CCNA2, TOP2A in the protein-protein interaction network.
Key role genes for lung cancer pathogenesis identified by PPI analysis
Key role genes for lung cancer pathogenesis identified by PPI analysis
Protein-protein interaction networks of lung cancer meta-signature genes. The node of the yellow-filled rectangle in the figure represented the 10 proteins with the largest number of effector proteins, and the related genes may have a greater impact on the pathogenesis of lung cancer.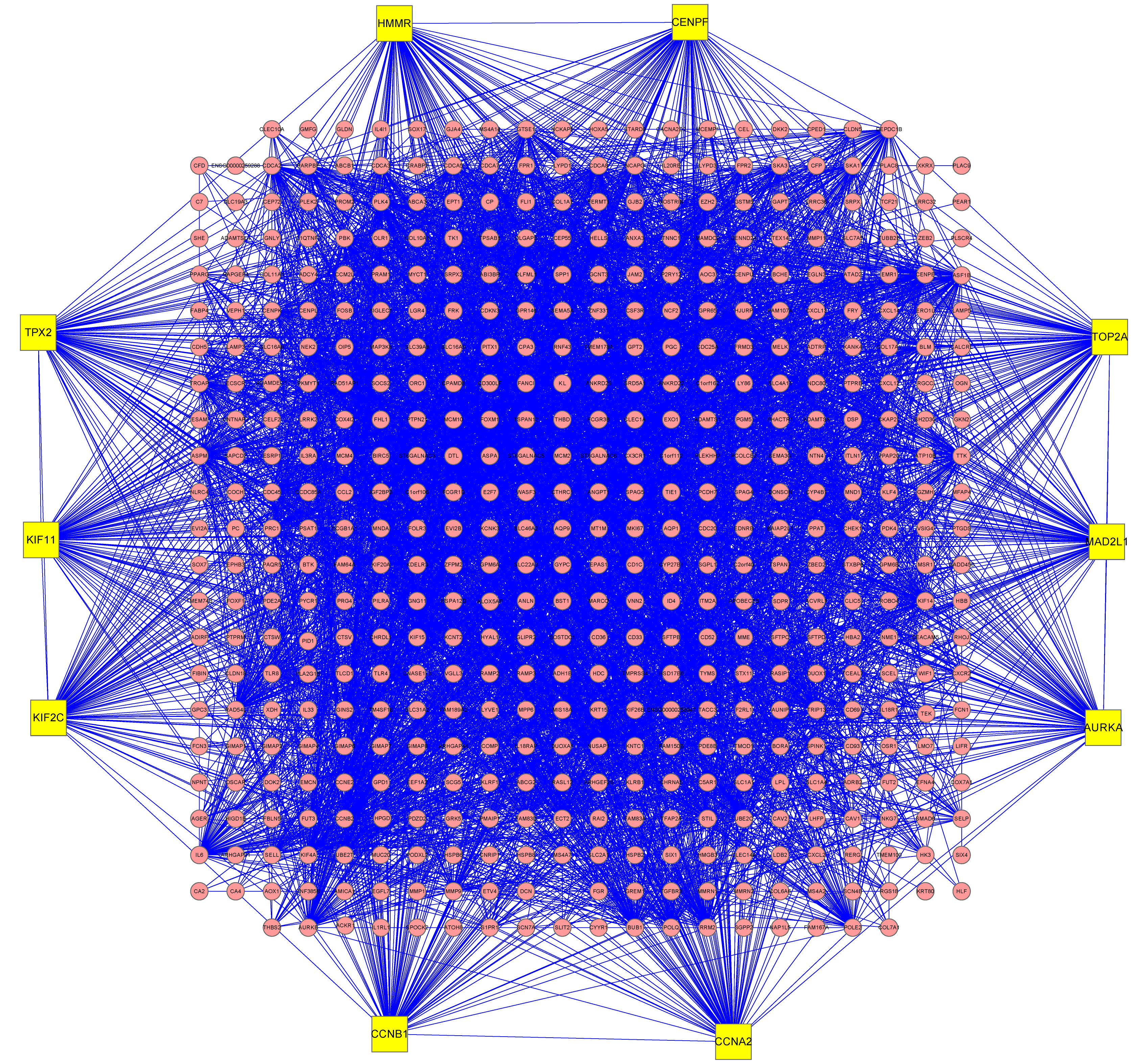
Furthermore, the transcription factor genes corresponding to DEGs were analyzed using the TfactS database, and the similarity and differences of transcription factor genes were compared corresponding to up- and down-regulation of DEGs. As shown in Fig. 5A, in the up-regulated DEGs, a total of 73 transcription factor genes formed 178 interactions with 49 genes, while in the down-regulation of DEGs, a total of 85 transcription factor genes formed 212 interactions with 62 genes. A total of 111 transcription factor genes regulated a total differentially expressed genes, and 47 transcription factor genes were shared by two types of target genes, accounting for 42.3% of all transcription factor genes.
Transcription factor analysis of differentially expressed genes up- and down-regulated expressed genes. A. Up-and-down adjustment of differentially expressed genes and comparison of unique transcription factors. The red background represents the number of transcription factors that up-regulate differentially expressed genes. Blue background represents the number of transcription factors that down-regulate differentially expressed genes. B. Transcription factor gene regulates the quantitative statistics of differentially expressed genes. C. The 10 transcription factor genes that regulate the largest number of genes.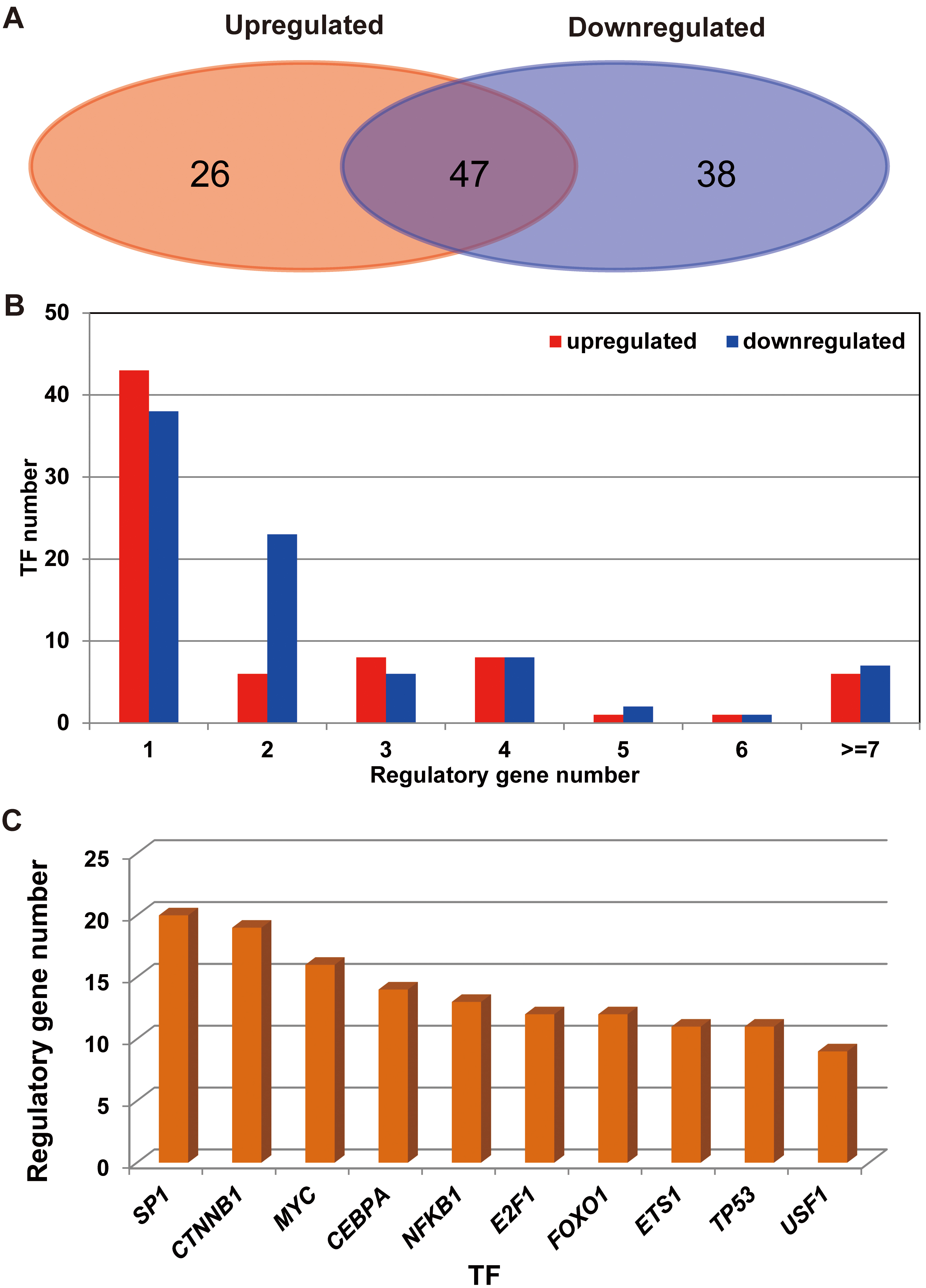
As shown in Fig. 5B, a higher proportion of transcription factor genes only regulated a single target gene. By statistically adjusting the number of target genes, it was found that the regulatory factors of DEGs such as SP1, CTNNB1, MYC, CEBPA, and NFKB1 were strong (Fig. 5C).
In this study, through meta and protein-protein interaction network analysis, ten important genes related to lung cancer incidence were identified. They were AURKA, CCNB1, KIF11, CCNA2, TOP2A, CENPF, KIF2C, TPX2, HMMR and MAD2L1. Among them, KIF11, belonging to the Kinesin-5 family, is related to kinds of mitotic microtubule functions, such as microtubule crosslinking, antiparallel microtubule sliding [27, 28]. Moreover, it has been found in cell proliferation and cancers including bladder cancer and pancreatic cancer [29, 30]. Thus, it may be used as a biomarker for chemotherapy. TOP2A, as a key nuclear protein, can control DNA topology [31, 32]. The overexpression of it happened in kinds of human cancers, including cervical carcinoma, breast cancer, and prostate cancer [33, 34]. KIF2C, as a member of the Kinesin family, affects spindle assembly and chromosome segregation during mitosis [35, 36]. The higher level of KIF2C is related to human cancer including gastric cancer, colorectal cancer, and breast cancer [37, 38]. TPX2, as a microtubule-related protein, has been reported in many studies, which are related to kinds of cancers including lung cancer, cervical cancer. HMMR has been shown to play an important role in the neoplastic progression. Moreover, the over-expression of it is associated with many human cancers and it can be used as an attractive target for inhibiting glioblastoma. MAD2L1 is an important mediator in the chromosomal control pathway [39]. Hyperexpression of MAD2L1 is correlated with poor prognosis of tumor patients [40]. The reports about the rest genes are few.
Interestingly, there was a down regulation in angiogenesis consistent across all data sets. The angiogenic process that is responsible for the support of tumor progression and metastasis represents one of the main hallmarks of cancer. Our hypothesis on why angiogenic factors would be downregulated in the context of lung cancer is that the downregulation of key angiogenic players could contribute to the control of extra thoracic invasion of cancer cells in human adenocarcinoma with predominant lepidic growth, which is consistent with previous report [41].
Additionally, it can be concluded that by statistically adjusting the number of target genes, it can be seen that the regulatory factors of differentially expressed genes such as SP1, CTNNB1, MYC, CEBPA and NFKB1 were stronger. SP1, as one of the members of the family of Sp/Kruppel – like factors, can be used as a transcription factor in various cancers and works as a transcriptional activator of target genes. By managing the transcription of various housekeeping genes, SP1 functions in kinds of biological processes, such as metabolism, cell cycle and cell death. Until now, it has been found that over 12,000 SP1 binding sites exist in human genes. SP1 play a key role in providing methods for cancer by regulating the functions of countless cellular genes [40]. CTNNB1 was reported to have a relationship with clonogenic cell growth, differentiation, and apoptosis. Previous studies showed that CTNNB1 is related to the oncogenesis of many human cancers, such as colorectal cancer and breast cancer. Also, it played a key role in tumor growth and metastasis. MYC belongs to the family of cellular oncogenes and functions as a pleiotropic transcription factors in various cellular processes. It has been studied that about 55% MYC is up-regulated. By many mechanisms such as gene duplications, somatic mutations, the stability of MYC can be increased [42]. MYC, as a frequently amplified proto-oncogene in human cancers, can adjust immune checkpoint genes and has been implicated in immune evasion by cancer cells. Meanwhile, MYC can be used as a potential target for new therapeutic strategies [42]. CEBPA, as a member of the leucine zipper family of transcription factors, can serve as a prognostic marker for various cancers. The family expresses in lots of tissues and cell types and controls many cellular processes, such as cellular proliferation and differentiation, inflammation and metabolism. NFKB1, as one of the members of the transcription factor family, is a possible gene for cancer susceptibility. Inappropriate activation NFKB1 is related to the development of various diseases and tumors. The pro-inflammatory NFKB1 plays an important role in inflammatory, while lung inflammation is the major pathogenetic feature for lung cancer.
Conclusions
In conclusion, in this study, a total of ten meta-signature genes in lung cancer and 10 transcription factors affecting the pathogenesis of lung cancer were identified. These 10 Meta-signature genes can be used as biomarkers for targeted prevention and treatment of lung cancer.
Footnotes
Acknowledgments
This study was supported by the innovative grant of the Department of Oncology, Tianjin Medical University General Hospital.
Conflict of interest
The authors declare that they have no competing interests.