
Abstract
BACKGROUND AND OBJECTIVE:
N6-methyladenosine (m6a) is the most abundant form of methylated modification in eukaryotic mRNA. However, the role of m6A-related genes in neuroblastoma (NB), one of the most common paediatric malignant tumours, is not well known. This study aimed to determine the prognostic role of m6A-related genes in neuroblastoma.
METHODS:
We analysed the expression of 20 published m6A methylation regulators in 498 patients with NB from the Gene Expression Omnibus database. To determine the independent prognostic factors, we used univariate Cox analysis, the least absolute shrinkage and selection operator (LASSO) regression. The multivariate Cox analysis was used to construct a prognostic risk prediction model. 120 NB tissues from “Therapeutically Applicable Research To Generate Effective Treatments” (TARGET ) database was used to test the prognostic value. Gene set enrichment analysis was performed to discover the potential biological function of the m6A signature.
RESULTS:
The risk prediction model consisted of five genes (METT14, WTAP, HNRNPC, YTHDF1 and IGF2BP2). The receiving operating characteristic curve showed the high exactitude of the risk model. Cox regression analysis revealed that the risk model was an independent prognostic factor of overall survival. These results were reproduced using another published independent dataset. Further functional enrichment analysis suggested the involvement of the 5-gene signature in several malignancies.
CONCLUSION:
The five m6A regulatory genes identified in this study enable clinical prognosis of NB and may serve as novel therapeutic targets for NB.
Introduction
Neuroblastoma (NB) is the most common extracranial solid tumour in children. It originates from the sympathetic ganglia and bilateral adrenal gland, with the highest incidence and mortality in infancy, accounting for 8%–10% of tumours in children [1, 2]. In the United States, NB occurs at a frequency of approximately 10.3 per million in children aged
It is well known that the phenotypic characteristics of tumours are regulated by genetic and epigenetic regulations. Of the latter, RNA modification is the most abundant, with currently more than 150 varieties [9]. N6-methyladenosine (m6a) is the most abundant eukaryotic mRNA modification and also the most thoroughly studied type [10]. High-throughput m6A sequencing studies revealed that m6A modifications comprising significant enrichment in 3’ UTRs near the stop codons are widespread at the transcriptome level, which affects thousands of mRNAs and non-coding RNAs [11, 12]. In general, m6A modification is mediated by a range of regulatory enzymes which consist of ‘writes’ (methyltransferases), METTL3, METTL14, RBM15, WTAP, VIRMA and ZC3H13, ‘erasers’ (demethylases) FTO and ALKBH5 and ‘readers’ YTHDC1, YTHDC2, YTHDF1, YTHDF2, YTHDF3, IGF2BP1, IGF2BP2, IGF2BP3, HNRNPC, HNRNPG/RBMX, FMR1 and EIF3 [13, 14, 15, 16].
Studies conducted over the past few decades have shown that m6A methylated modification play an important role in several major physiological processes such as brain development, spermatogenesis, haematopoietic stem and progenitor cell specification, control of the circadian clock and so on [17, 18, 19, 20]. More recently, the impact of dysregulated m6A modification (mediated by regulatory enzymes) on various cancers has been extensively studied [16]. However, evidence remains insufficient, particularly in paediatric malignant tumours.
In this study, we systematically analysed twenty widely reported m6A regulators expression and clinical characteristics in 498 NB tissues from the Gene Expression Omnibus (GEO) dataset, GSE79711. We found that the abnormal expression of these genes was closely related to NB prognostic risk factors, and also identified a five-gene signature that may be a prognostic marker of NB.
Materials and methods
Patient datasets and m6A regulators
The expression data and clinical information were retrieved from the GEO and Therapeutically Applicable Research To Generate Effective Treatments (TARGET) databases. The RNA sequencing dataset, GSE49711, of 498 patients with NB downloaded from the GEO database was used as the training set. The RNA sequencing dataset of 120 patients with NB from TARGET database was used as the testing set. All the clinical characteristics of patients with NB in the two datasets are summarised in Supplementary Table S1 and Table S2. The 20 m6A methylation regulators were identified from published studies [13, 14, 15, 16].
Bioinformatic and statistical analysis
To inspect the role of m6A methylation regulators in NB development, we applied R package of limma [21] to analyse their differential expression in each clinicopathological characteristic. A
Expression and interaction of m6A methylation regulators in neuroblastoma. (A, B) Differential expression of m6A methylation regulators between subgroups stratified by progression and COG risk (Children’s Oncology Group risk stratification). N, non-progression; P, progression; *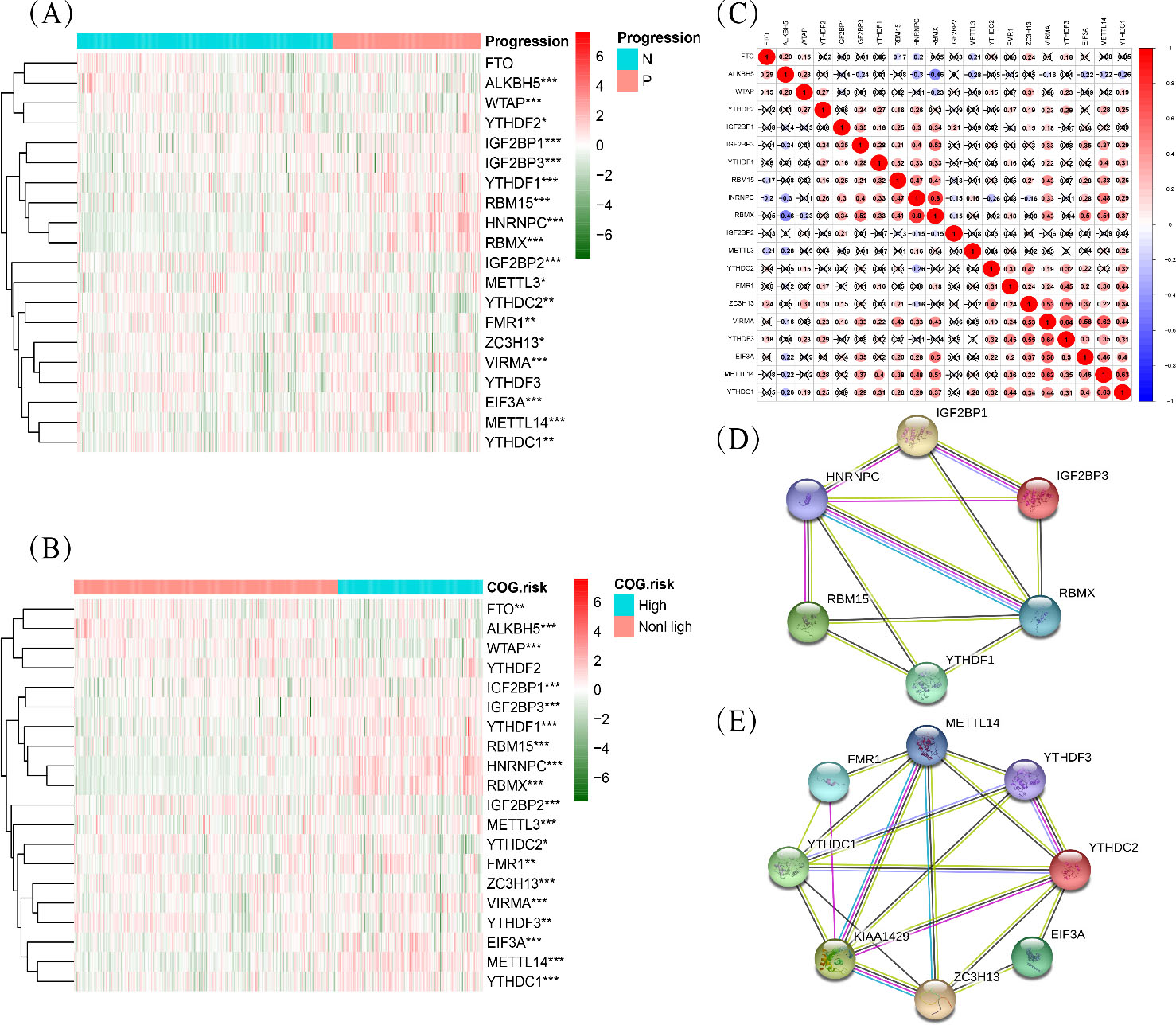
To determine the prognostic value of m6A regulators, univariate Cox regression analysis was applied to determine the association between m6A regulators expression and overall survival in the training set. From the regulators with significant results, we used the least absolute shrinkage and selection operator (LASSO) regression [24] and multivariate Cox regression to screen and establish a prognostic signature model. The time-dependent receiver operating characteristic (ROC) curve was used to evaluate the exactitude of the risk model.
where
Univariate Cox regression (A) and LASSO regression (B, C) analysis evaluating independently predictive ability of m6A methylation regulators for overall survival of NB patients.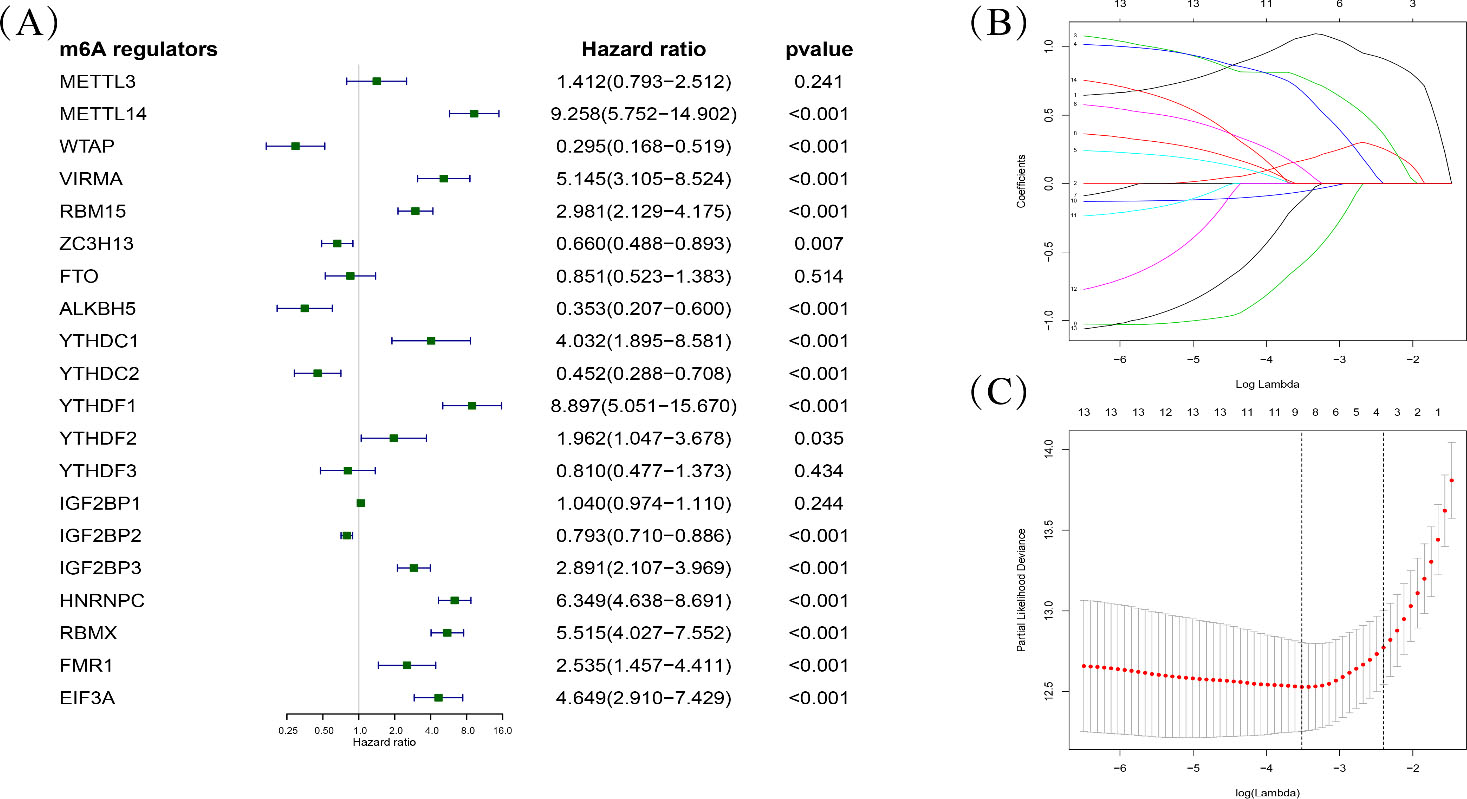
Specific expression of m6A methylation regulators in NB
To show that m6A methylation regulators play an important role in the development of neuroblastoma, we comprehensively analysed their differential expression in two vital clinicopathological characteristics, tumour progression and COG risk group. Expression patterns presented as heatmaps (Fig. 1A and B) indicated a close correlation between m6A regulators and prognostic risk of tumours. Additionally, the genetic change frequencies of the 20 m6A regulators were significantly low in NB (Supplementary Fig. S1). These results indicated that the change in expression of m6A regulators was not due to genetic mutations and copy number changes. To further understand the interactions among the 20 m6A regulators, we analysed their expressed correlation (Fig. 1C). As can be seen from the correlation-plot, strong co-expression relationships between YTHDC2, FMR1, ZC3H13, VIRMA, YTHDF3, EIF3A, METTL14 and YTHDC1. Besides, IGFBP1, IGFBP3, YTHDF1,RBM15, HNRNPC and RBMX also displayed interactions with each other. These results were also reported in STRING database (Fig. 1D and E).
Development of the prognostic risk model
To identify the prognostic m6A methylation regulators, univariate Cox proportional hazard regression analysis was performed on each regulator in the training set (Fig. 2A). A total of 14 genes were significantly related to overall survival (
M6A methylation regulators significantly associated with overall survival of NB patients
M6A methylation regulators significantly associated with overall survival of NB patients
NB, neuroblastoma; HR, hazard ratio; 95% CI, 95% confidence interval.
To investigate the prognostic value of the five-gene signature, the differences in clinical characteristics between divided into high-risk and low-risk patients with NB based on the median risk score were evaluated. The heatmap showed that all the clinical risk factors, including age, MYCN status, COG risk group, INSS stage, histology and tumour progression, were significantly different between the high-risk and low-risk groups, and the survival state associated with risk score was also presented (Fig. 3A). Kaplan-Meier curve analysis, which evaluate the survival differences between the two groups (Fig. 3B), showed that the 5-year overall survival rates of high-risk and low-risk groups were 57.0% (95% CI: 50.8%, 64.1%) and 98.1% (95% CI: 96.2%, 100%), respectively. The log-rank test indicated overall survival rates were significantly different between the two risk groups (
Evaluation of the risk predictive model in GSE49711 dataset. (A) The distribution of clinical characteristics, m6A regulators’ expression level, patients’ survival status and risk score between high- and low-risk group. COG, Children’s Oncology Group; INSS, International Neuroblastoma Staging System; Y, yes; N, no; Amp, amplified. (B) Kaplan-Meier curve for overall survival of NB patients of high- and low-risk groups. (C) Receiver operating characteristic (ROC) analysis for 5-year overall survival prediction of the risk model. (D–I) Comparation of risk scores in subgroups stratified by age, MYCN status, COG risk, histology, progression and INSS stage. *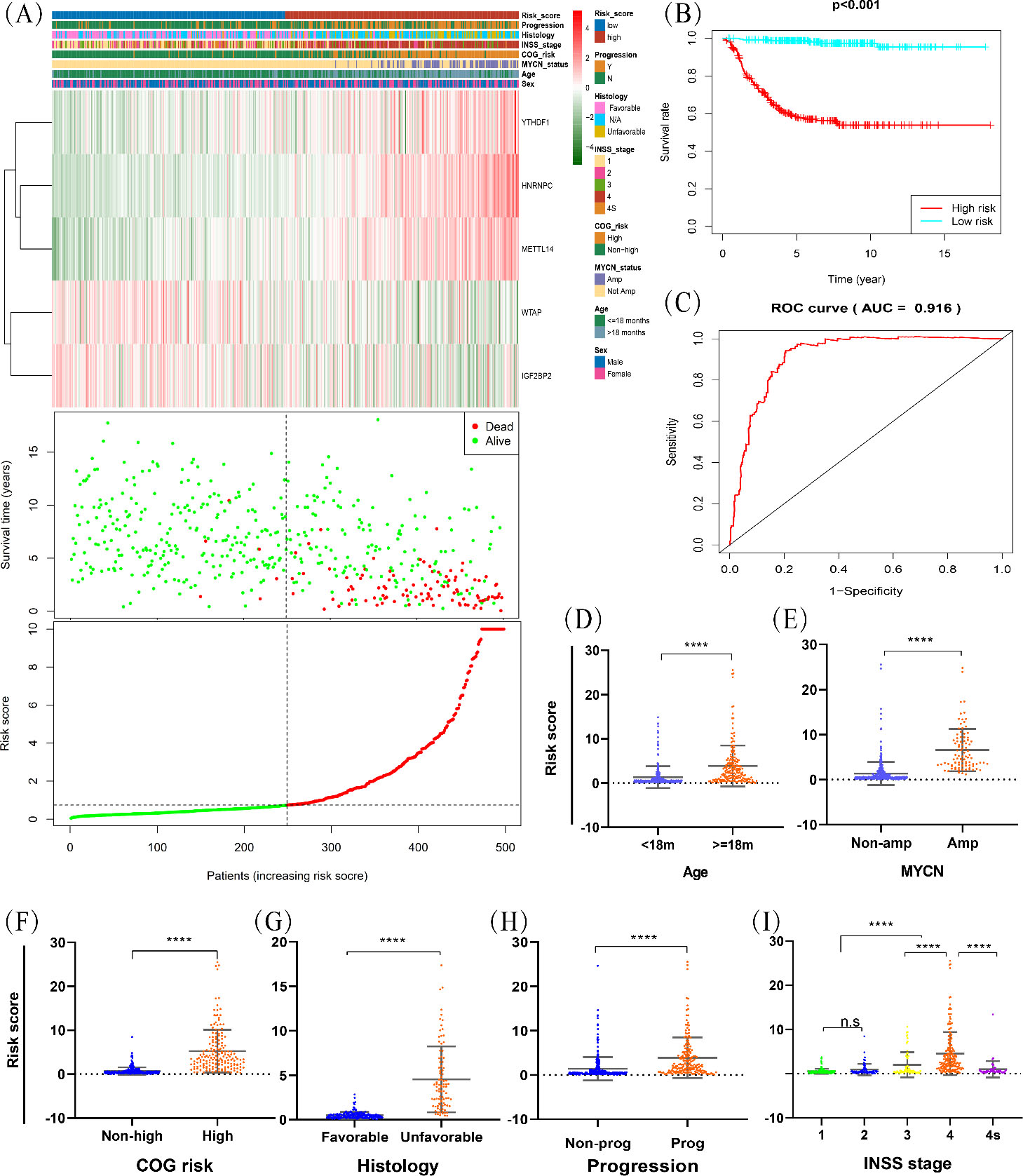
Investigation of the differences in risk score according to clinicopathological features showed significant differences between subgroups stratified by age, INSS stage, MYCN status, tumour progression, histology and COG risk (Fig. 3 D–I). These results reinforce the stability of the prognostic risk prediction model and reliability of the training set. Univariate Cox regression analysis to assess whether the risk gene signature was independent of other clinicopathological features suggested that age, MYCN status, COG risk, INSS stage and risk score were associated with overall survival (Fig. 4A). Including these variables into the multivariate Cox regression analysis showed that MYCN status, COG risk, INSS stage and risk score remained independent prognostic indicators for overall survival (Fig. 4B).
Univariate (A) and multivariate (B) Cox regression analyses in overall survival of NB patients among the gene signature (risk score) and clinicopathological factors. COG, Children’s Oncology Group; INSS, International Neuroblastoma Staging System.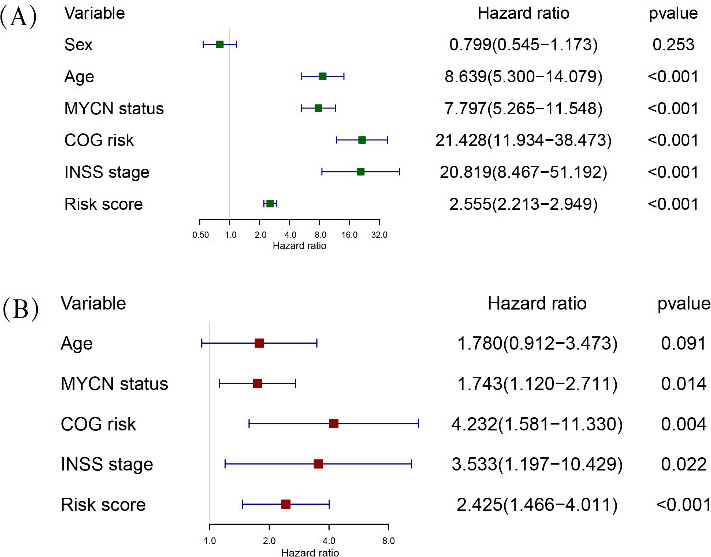
Evaluation of the risk predictive model in TARGET dataset. (A) Kaplan-Meier curve for overall survival of NB patients of high- and low-risk groups. (B) Receiver operating characteristic (ROC) analysis for 5-year overall survival prediction of the risk model. (C, D) Univariate and multivariate Cox regression analyses in overall survival of NB patients among the gene signature (risk score) and clinicopathological factors. COG, Children’s Oncology Group; INSS, International Neuroblastoma Staging System; MKI, mitotic karyorrhexis index.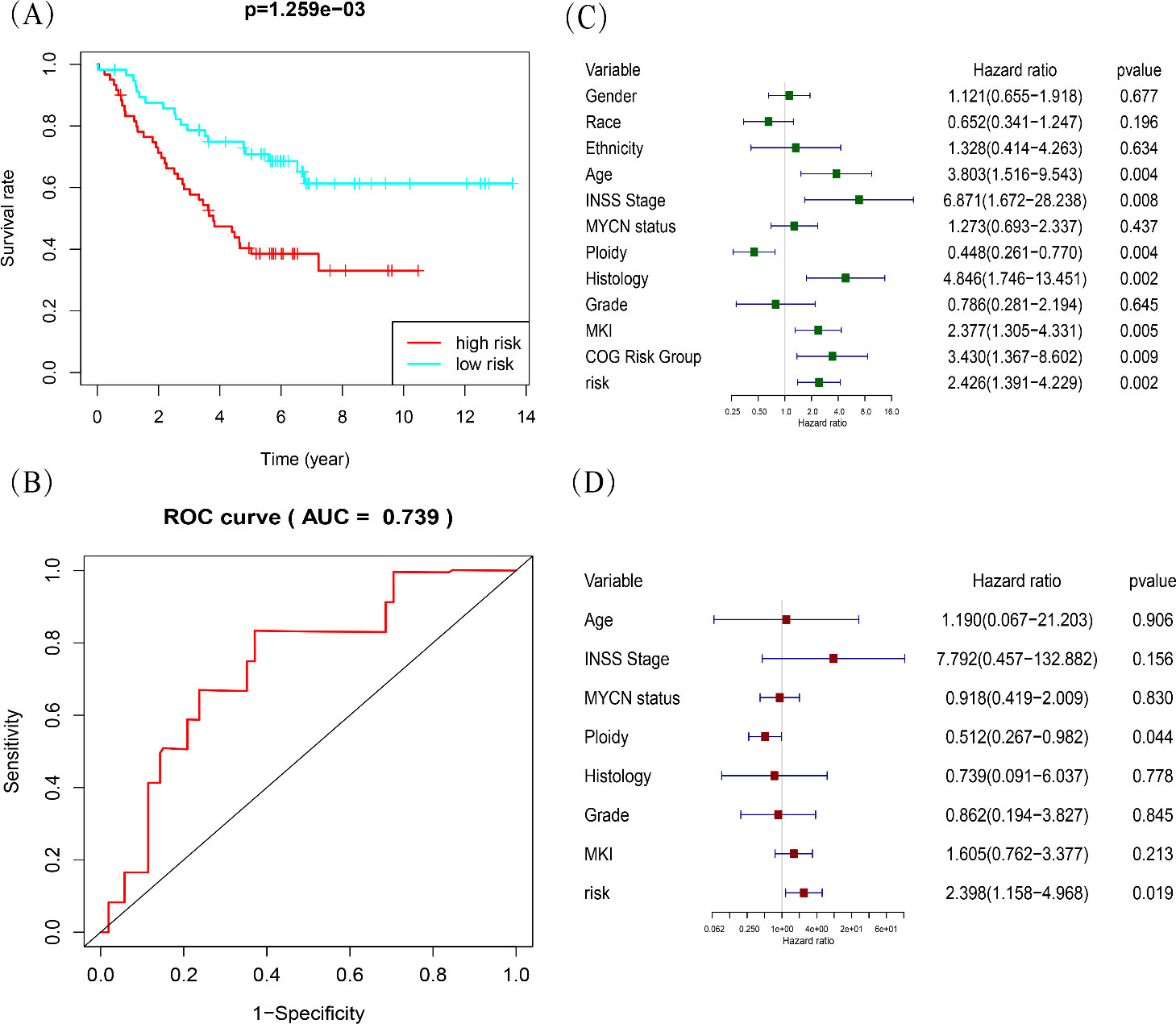
Gene set enrichment analysis (GSEA) of the high-risk group.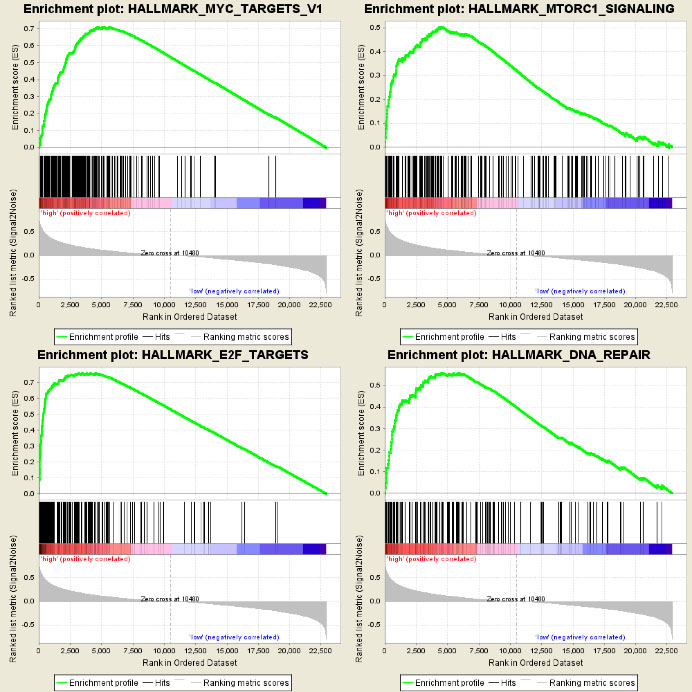
To validate the robustness of our risk model, the TARGET database was used as the external validation dataset to test the prognostic value. The risk score for each patient was calculated according to our risk model. Patients were divided into high-risk and low-risk groups based on the median risk score. Analysis of Kaplan-Meier curves to determine the 5-year overall survival of the two groups (Fig. 5A) revealed that patients with high-risk scores showed poor overall survival (OS, Log-rank test
Functional enrichment analysis
Using GSEA to identify associated biological processes and pathways in the GSE49711 dataset revealed several cancer-related networks, including mTORC1 signalling (NES
Discussion
A systematic analysis of twenty widely reported m6A regulators expression and clinical characteristics in 498 NB tissues revealed that the regulators of m6A methylation, the most abundant form of methylated modification in eukaryotic mRNA, play a significant role in the prognosis of NB. Traditional stratified treatment strategies have made progress in improving overall survival of patients with NB [6, 25], although the outcomes of patients with highly aggressive NB remain unsatisfactory. Children with NB, the most common paediatric extracranial solid tumour, respond differently to therapy although their tumours share similar clinicopathologic factors, warranting the urgent need for identification of novel molecular prognostic indicators beyond traditional clinicopathologic factors for NB. Epigenetic abnormalities are closely associated with malignant behaviours in paediatric cancer [26, 27, 28, 29]. In the past few decades, there were a large number of intensive studies on DNA methylation, but little on RNA methylation.
By analysing high-throughput transcriptome data, we found that the differential expression of m6A regulators was significantly related to NB malignancy. Based on the differential expression of the m6A regulators, we identified a five-gene signature that predicted different overall survival between high-risk and low-risk patients with NB using the risk prediction model. The prognostic value of the risk model was validated in TARGET, an independent dataset. However, compared with the training set, overall survival was low because the majority of the dataset comprised stage 4 patients leading to a smaller difference between high-risk and low-risk patients with NB. Nevertheless, the advantage of this approach was the ability to predict overall survival in patients at the high-risk stage using this risk model. By GSEA functional enrichment analysis, we found that the high-risk group was closely correlated with hallmarks of tumour malignancy, including mTORC1 signalling, MYC targets, E2F targets and DNA repair.
M6A regulators are classified by function as writers, erasers and readers, which regulate RNA translation, mRNA splicing, transport, localisation and stability [13, 30, 31]. Our risk model consisted of two writers (METTL14, WTAP) and three readers (YTHDF1, HNRNPC, IGF2BP2). Studies have shown that the roles of m6A methylation regulators differ with tumour types. METTL14 promotes tumorigenesis by upregulating expression of MYC and MYB in acute myelocytic leukaemia (AML), but acts as a tumour suppressor by inactivating AKT in endometrial cancer [32, 33]. We showed that high expression of METTL14 significantly correlated with low overall survival of patients with NB, and the high-risk group was highly enriched in MYC targets, suggesting that METTL14 exerted its potential functions through MYC-associated pathways. WTAP promotes proliferation, invasion, metastasis and chemo-resistance of multiple cancers, including AML, pancreatic cancer, renal cell carcinoma, colorectal cancer etc. [34, 35, 36, 37]. Not surprisingly, the low expression of WTAP correlated to low overall survival of patients with NB in this study.
The reader HNRNPC has the largest weight in the risk model; this is consistent with a recent study which identified that the overexpression of HNRNPC played a critical role in alternative cleavage and polyadenylation for metastatic colon cancer cells [38]. Our study confirmed that overexpression of YTHDF1 was highly associated with low overall survival of patients with NB. Others have shown that YTHDF1 increased translation efficiency of m6A-modified mRNA and was associated with poor cancer prognosis [39], and reduced the therapeutic efficacy of PD-L1 checkpoint blocker through its function as an anti-tumour immunity factor [40]. Bell et al., used IGF2BP paralog-specific custom-made monoclonal mouse antibodies and western blotting to investigate expression of IGF2BPs in aggressive neuroblastoma and showed increase of IGF2BP1 in stage 4 tumours and decrease in IGF2BP2, compared with stage 1–3 tumours [41], which were consistent with our findings. Although Cheng et al. recommended IGF2BP2 as a candidate tumour suppressor through pan-cancer analysis of homozygous deletions [42], others reported overexpression of IGF2BP2 in multiple cancers and correlated it with poor survival [43, 44]. These results indicated diversity in function of m6A methylation regulators in different tumours, depending on their target genes.
Considerable attention has been devoted to investigating the role of m6A methylation in cancers. We demonstrated through transcriptomic analysis that expression of m6A methylation regulators correlated with malignant progression of NB. Nevertheless, several limitations should be noted. At present, studies on m6A methylation are relatively few and therefore, improvement in the accuracy of risk models requires the inclusion of additional m6A regulators. A correlation between m6A regulators and clinicopathologic factors was investigated, but a causal relationship was not established. Future biological experiments are required. Also, the gene signature identified as the independent prognostic factor in this study was based on two published datasets with certain differences in clinical characteristics, warranting the need for additional datasets to validate our findings.
In conclusion, our study revealed a risk model, composed of five m6A regulatory genes, that is of considerable significance for overall survival of patients with NB. The high-risk score of the model correlated with malignant pathological features. Since the prognostic value of the risk model was independent of traditional clinicopathologic characteristics, the regulatory genes identified in this model may serve as novel biomarkers for the prognosis of patients with NB, enabling clinicians to adopt individualised therapeutic regimens. Moreover, the findings also provide an impetus for basic medical research of m6A methylation in neuroblastoma.
Footnotes
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.